缺血性卒中后,缺血核心和梗死灶周围区的损伤影响患者预后。微胶质细胞立即对缺血性打击做出反应,引发免疫炎症,在卒中后的细胞损伤中发挥着重要作用。然而,微胶质细胞的多样性和涉及的机制仍然不清楚。
我们首先对三个时间点的大鼠大脑进行了
scRNA-seq
和空间转录组学(
ST
),以确定与卒中相关的微胶质细胞亚簇及其空间分布。此外,通过
RNAscope
和免疫荧光技术在大鼠中验证了微胶质细胞亚群特异性标记基因的表达和不同微胶质细胞亚群的定位。进行基因集变异分析(
GSVA
)以揭示微胶质细胞亚群的功能特征。此外,使用调查通路分析(
IPA
)来探索微胶质细胞亚群的上游调控因子,通过免疫荧光、
RT-qPCR
、
shRNA
介导的基因敲除和定向代谢组学进行确认。最后,在特定微胶质细胞亚群操纵后,评估大鼠
MCAO
模型中的梗死灶大小、神经功能缺陷和神经元凋亡。
我们在
MCAO
大鼠大脑中发现了与卒中相关的微胶质细胞亚群。我们还确定了这些微胶质细胞亚群的新标记基因,并根据它们的空间分布将这些细胞定义为缺血性核心相关(
ICAM
)和梗死灶相关(
IPAM
)的微胶质细胞。
ICAM
由损伤相关分子模式诱导,可能由糖酵解提供动力,并表现出增加的促炎细胞因子和趋化因子产生。
BACH1
是驱动
ICAM
生成的关键转录因子。相反,梗死灶中富含的糖皮质激素可能触发
IPAM
的形成,这些细胞可能由柠檬酸循环和氧化磷酸化提供动力,并以中等的促炎反应、抗炎代谢特征和髓鞘营养特性为特征。
ICAM
可能引起过度神经炎症,加剧脑损伤,而
IPAM
可能表现出神经保护特性,对于梗死灶中细胞的稳态和存活可能至关重要。我们的研究结果为以特定微胶质细胞亚群为靶点的治疗策略提供了生物学基础,成为缺血性卒中的潜在治疗策略。
该研究于
2023
年
12
月发表在《
Genome medicine
》,
IF
:
12.3
。
技术路线:
结果
:
1
、细胞类型的鉴定
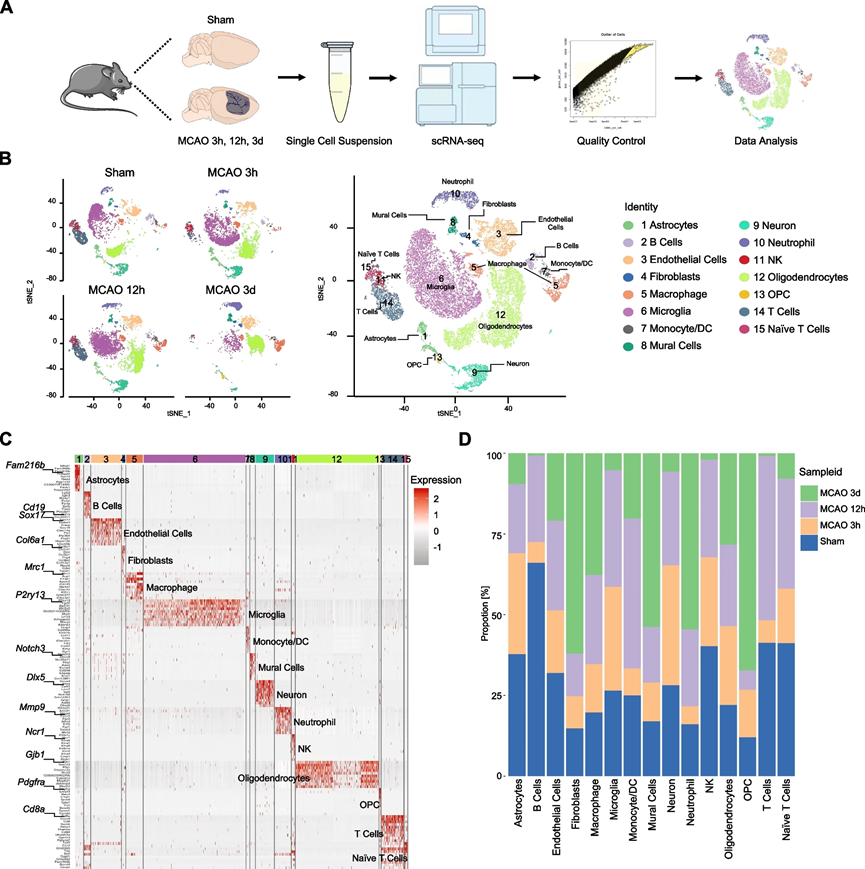
为了全面表征缺血性卒中后缺血半球主要细胞类型的变化,我们在经历短暂性大鼠中脑动脉阻塞(
MCAO
)的大鼠的脑样本中,采用
scRNA-seq
技术,在再灌注后的不同时间点(
3
小时、
12
小时和
72
小时)以及模拟大鼠上进行了细胞分离(图
1A
)。
3
小时和
12
小时的时间点代表了缺血性卒中的亚急性阶段,而
72
小时的时间点被选为进行急性阶段变化的转录分析。总共评估了
39,333
个细胞,每个细胞平均含有
1672-8800
个独特的分子标识符(
UMIs
),经过严格的
CellRanger
质量控制。在四个组中,每个细胞的平均基因数为
901-2636
。
接下来,我们进行了无监督聚类,并根据经典基因标记表达进行了细胞类型的注释。如
t-
分布随机邻居嵌入(
t-SNE
)图中所示,鉴定了
15
种不同的细胞类型,包括
B
细胞、内皮细胞、壁细胞、自然杀伤细胞(
NK
细胞)、单核
/
树突细胞(
DC
细胞)、成熟型寡能神经胶质细胞(
OPC
)、
T
细胞、星形胶质细胞、成纤维细胞、巨噬细胞、微胶质细胞、神经元、中性粒细胞、寡能神经胶质细胞和原始
T
细胞(图
1B
)。值得注意的是,微胶质细胞、寡能神经胶质细胞和内皮细胞占细胞总数的大多数。我们确定了每种细胞类型的所有标记基因,并在热图中显示了前
10
个标记基因(图
1C
)。
与先前的发现一致,缺血性打击后浸润的巨噬细胞和中性粒细胞数量逐渐增加,表明血脑屏障功能失调。此外,
OPC
的数量在手术后
72
小时显著增加,表明中风后
OPC
的增殖,这与先前的研究结果一致。我们还观察到在缺血后星形胶质细胞数量逐渐减少,而在
72
小时时微胶质细胞显著减少,这可能归因于急性缺血性损伤(图
1D
)。总体而言,这些数据提供了对缺血半球细胞组成的基本描述。
2
、缺血诱导两种类型的小胶质细胞亚簇
考虑到微胶质细胞在大脑中的重要性和微胶质细胞转录组的高质量,我们将焦点放在了单细胞水平的微胶质细胞上。我们在不同时间点确定了微胶质细胞的差异表达基因(
DEGs
)。与先前的研究结果一致,微胶质细胞
DEGs
的
KEGG
分析表明,在缺血性卒中的急性阶段,微胶质细胞的吞噬和炎症能力增强,代谢发生改变。此外,微胶质细胞被分为四个亚群进行进一步的分析(图
2A
)。根据亚群比例图,模拟组中
94%
的微胶质细胞属于亚群
2
。此外,微胶质细胞的稳态基因(
Tmem119
、
P2ry12
、
P2ry13
、
Csf1r
、
Cx3cr1
、
Hexb
)在亚群
2
中高度表达,表明亚群
2
可能由稳态微胶质细胞组成。亚群
4
(
465
个细胞)包括相对较少数量的微胶质细胞,在模拟和
MCAO
大鼠之间变化不大;因此,亚群
4
的表型和功能特征未进一步研究。亚群
1
和亚群
3
占据了
MCAO
组中微胶质细胞的大多数。这些发现将亚群
1
和亚群
3
定位为缺血性卒中急性阶段的关键角色,并提出了它们如何协调缺血性攻击适应的问题。
进行差异基因表达分析以识别亚群特异性标记基因。大多数高表达的基因也是亚群特异性的,表明微胶质细胞亚群具有独特的基因表达模式(图
2B
)。亚群
1
表达了先前未报道的标记基因(例如,
Pik3ip1
、
Cd300lf
、
Gpr65
、
Ms4a6c
、
Cep152
、
Ddit4
、
Zbtb16
、
Abhd15
、
Bmf
和
Arhgap24
)。在亚群
3
中,
Lgals3
、
Plau
、
Srxn1
、
Ankrd33b
、
Gas2l3
、
Nes
、
Edn1
、
Fam129b
、
Vat1
和
Fmn1
的表达上调。前
10
个亚群特异性标记基因可可靠地识别微胶质细胞亚群
1
和亚群
3
。此外,疾病相关微胶质细胞(
DAM
)曾被确定为与阿尔茨海默病(
AD
)相关的一种独特的微胶质细胞亚型。我们检查了我们数据集中已确定的
DAM
标记基因的表达水平。在四个亚群中,一些基因(例如
Tyrobp
、
Trem2
、
Apoe
、
Timp2
和
B2m
)的表达保持不变。在亚群
3
中,一些基因的表达上调,例如
Lgal3
、
Fth1
、
Cstb
、
Csf1
、
Cd63
、
Cd9
、
Ccl3
和
Lilrb4a
,而与稳态微胶质细胞(亚群
2
)相比,亚群
1
中这些基因的表达保持不变,表明亚群
3
可能表现出
DAM
样的特征。
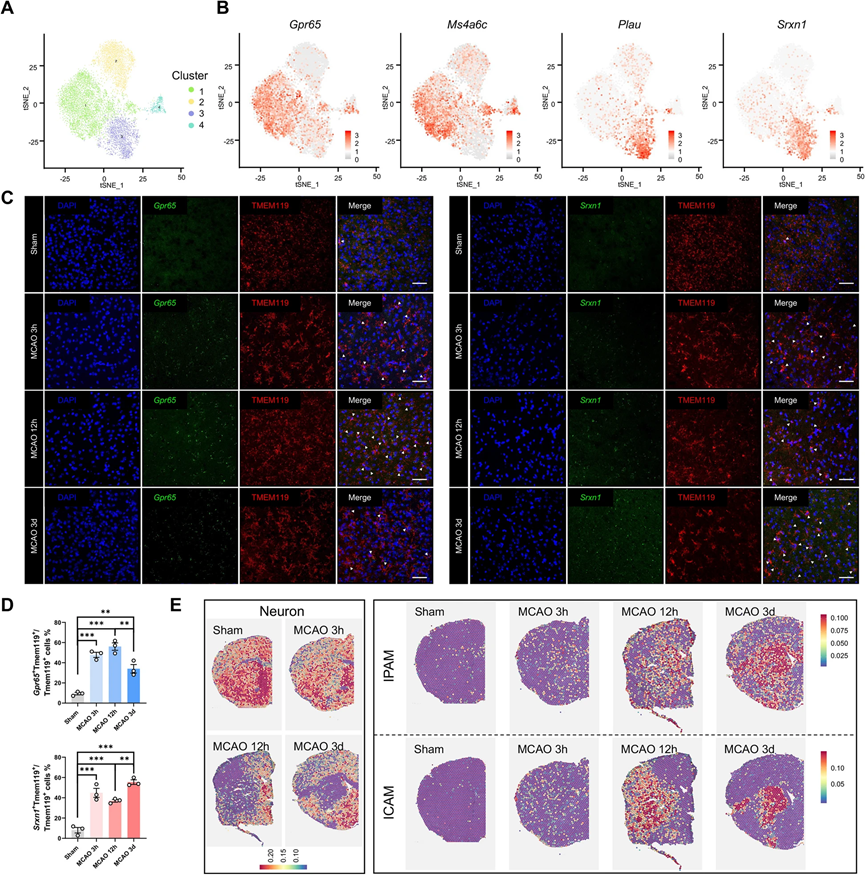
为了确定亚群
1
和亚群
3
是否具有时间特性,我们使用
RNAscope
(
ACD
,
Newark
,
CA
,
USA
)进行了
RNA-
蛋白共染色,使用亚群
1
特异性标记基因
Gpr65
和亚群
3
特异性标记基因
Srxn1
标记不同的微胶质细胞群体(图
2C
)。在再灌注后,亚群
1
和亚群
3
均作为对缺血性打击的迅速反应者(图
2D
)。为了确认亚群
1
和亚群
3
是否也存在于其他关于缺血性卒中的单细胞
RNA
测序研究数据中,我们重新分析了几个现有的缺血性卒中后微胶质细胞的数据集(
GSE174574
,
GSE197731
,
GSE227651
)。与我们的结果一致,亚群
1
和亚群
3
在它们的
t-SNE
微胶质细胞图中可以清晰地区分出来。
为了确定亚群
1
和亚群
3
是否与梗死核心存在任何空间关系,我们使用了空间转录组学(
ST
)来研究模拟和
MCAO
大鼠的脑切片。尽管
ST
几乎无法达到单细胞分辨率,但仍然可以通过计算与
scRNA-seq
数据相关的亚群特异性标记基因的平均表达来获得脑切片中不同细胞亚群的大致位置。为了定义缺血性核心,我们还查询了在脑切片上每个点的下调表明神经元丧失的神经元基因。引人注目的是,亚群
3
标记基因占据了缺血性核心,而亚群
1
标记基因则环绕着缺血病灶在缺血后
12
小时(图
2E
)。因此,我们将亚群
1
定义为缺血性梗死后的微胶质细胞(
IPAM
),将亚群
3
定义为缺血性梗死核心相关的微胶质细胞(
ICAM
)。
为了验证这些发现,我们在免疫荧光实验中使用
ICAM
特异性的
LGALS3
作为
ICAM
标记,而选择
PIK3IP1
作为
IPAM
标记。在
12
小时和
72
小时时,
ICAM
标记在缺血性核心中显著富集。
IPAM
标记在
12
小时时在缺血性梗死灶周围的位置也得到了确认。此外,
ICAM
特异性标记(
Lgals3
)和
IPAM
特异性标记(
Pik3ip1
、
Gpr65
和
Ms4a6c
)也在
ST
中可视化显示。我们还根据图
2E
将脑切片分为两个区域(缺血性核心和梗死周围区)。在缺血性核心中上调的基因被定义为缺血性核心基因(
ICGs
),而在梗死周围上调的基因被称为缺血性梗死周围基因(
IPGs
)。与
ICGs
重叠的
ICAM
标记更多,与
IPGs
重叠的
IPAM
标记也更多,进一步验证了
ICAM
和
IPAM
的空间分布。因此,我们的数据揭示了两个在缺血性卒中应答中涉及的在空间上和转录上具有不同特性的微胶质细胞亚群。
3
、
ICAM
和
IPAM
表现出截然不同的特征
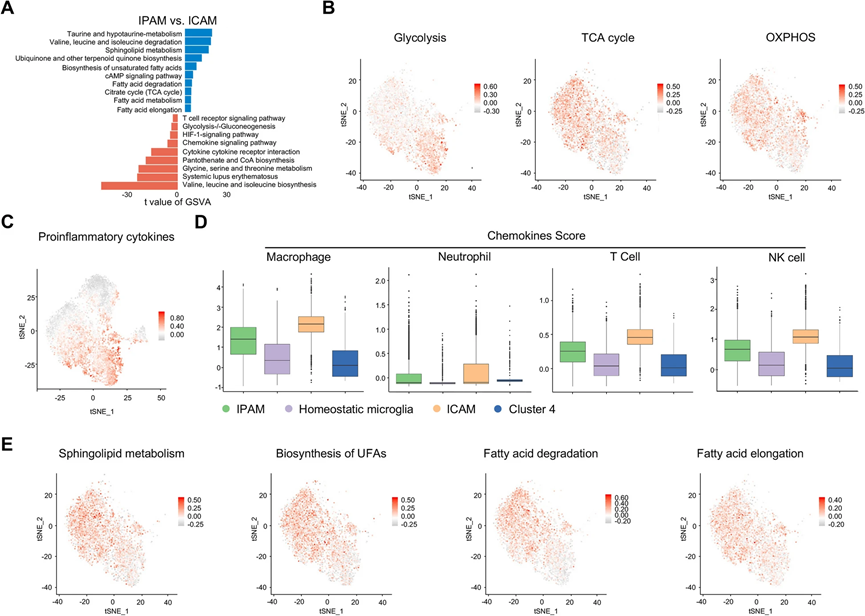
对于功能实验,我们对
IPAM
和
ICAM
进行了基因集变异分析(
GSVA
)(图
3A
)。我们发现
ICAM
可能在代谢上更依赖于糖酵解,而
IPAM
可能依赖于三羧酸循环(
TCA
)和氧化磷酸化(
OXPHOS
),通过使用
Seurat
中的
AddModuleScore
计算与感兴趣的代谢途径相关的基因的表达来证实(图
3A
、
B
)。一致地,
“
氧化磷酸化
”
和
“
柠檬酸循环
”
在
IPG
中也富集,如
KEGG
分析所示。
ICAM
中
“
断裂
”
的
TCA
循环也可能导致中间产物水平的增加,如
ICAM
中增强的泛酮酸和辅酶
A
生物合成所示。此外,在
IPAM
中观察到
AMPK
和
cAMP
信号通路的上调,表明
IPAM
使用了完全不同的能源来源(图
3A
)。此外,微胶质细胞的代谢重编程依赖于
HIF-1α
通路,
ICAM
中
HIF-1α
通路也高度表达(图
3A
)。
我们进一步发现相对于其他亚群,
ICAM
中存在一些促炎和炎症响应基因(
Il1a
、
Il1b
、
Il6
、
Il18
、
Tnf
、
Hmox1
、
Ptgs2
)的表达上调(图
3C
)。此外,巨噬细胞和中性粒细胞在
MCAO
后
12
小时开始浸润大脑,并在
72
小时时聚集在梗死核心,这表明这些外周免疫细胞在空间上与
ICAM
存在重叠。由于微胶质细胞是缺血性卒中中各种趋化因子的主要来源,我们检查了
ICAM
和
IPAM
中驱动不同外周免疫细胞招募的一系列趋化因子的表达。因此,
ICAM
中富集了不同外周免疫细胞中相关趋化因子基因集的表达(图
3D
)。具体而言,与
IPAM
相比,
ICAM
中
Ccl2
、
Ccl3
、
Ccl4
、
Ccl5
、
Cxcl2
和
Cxcl16
的表达水平更高。此外,
KEGG
分析一致表明,促炎信号通路(
“TNF
信号通路
”
、
“IL-17
信号通路
”
、
“MAPK
信号通路
”
)在
ICG
中显著富集。这些发现表明由
ICAM
介导的过度促炎和趋化反应,提示
ICAM
可能在缺血性卒中急性阶段加速组织损伤,具有不利影响。
此外,与
ICAM
相比,
GSVA
分析显示,
IPAM
中与氨基酸、脂质和碳水化合物相关的几个代谢途径显著富集(图
3A
)。支链氨基酸(
BCAAs
)如缬氨酸、亮氨酸和异亮氨酸的水平在
ICAM
中升高,而在
IPAM
中
BCAAs
降解被增强(图
3E
)。此外,
IPAM
中其他氨基酸的代谢,如牛磺酸,也富集(图
3A
)。根据
GSVA
数据,脂质代谢在
IPAM
中也很活跃,包括鞘脂代谢、主要胆酸生物合成、不饱和脂肪酸生物合成、脂肪酸降解和延长,通过
AddModuleScore
确认(图
3A
,
3E
)。此外,
IPAM
中富集的代谢途径(
“BCAAs
降解
”
、
“
不饱和脂肪酸生物合成
”
、
“
脂肪酸降解
”
、
“
脂肪酸延长
”
、
“
鞘脂代谢
”
)在
ST
中也显示了一个梗死周围的分布。
由于脂质代谢产物是髓鞘的重要组成部分,
IPAM
可能通过供应脂质组分来补偿寡突细胞。此外,我们使用
CellPhoneDB
分析了
IPAM-
寡突细胞相互作用中的配体
-
受体相互作用。已经确定了几对配体
-
受体对,其中
GRN-SORT1
是其中最显著的一对。为了测试
GRN
是否具有寡突细胞营养的潜力,我们用重组小鼠
GRN
(
rmGRN
)处理寡突细胞,发现
rmGRN
显著促进了
MAG
的表达。
总的来说,这些独特的特征至少在一定程度上塑造了微胶质细胞的应答,并为理解微胶质细胞亚群在对缺血刺激的响应中不同的激活状态提供了新的视角。
4
、
M1/M2
二分法无法对缺血后的小胶质细胞亚簇进行分类
在历史上,基于体外刺激,微胶质细胞曾经简单地被分类为促炎性的“
M1”
和抗炎性的
“M2”
。然而,根据我们的数据,
M1
极化评分的增加总是伴随着不同时间点上升的
M2
极化评分(图
4A
)。我们想知道我们的无监督聚类是否与这种表型分类相对应。为了测试这个假设,我们分别在
IPAM
和
ICAM
上执行了
M1/M2
极化评分。然而,
ICAM
的
M1
和
M2
极化评分都高于
IPAM
(图
4B-D
)。此外,
IPAM
和
ICAM
在缺血损伤后的不同时间点上展现出相同的
M1/M2
极化改变趋势(图
4E-F
)。
接下来,我们还进行了伪时轨迹分析,并观察到在缺血后出现了明显的分歧轨迹。为了阐明这两个分支是否与
M1-
和
M2-
表型微胶质细胞一致,我们检查了不同轨迹中
M1/M2
标记基因的表达。在这两个分支之间,
M1/M2
标记基因的表达仍然没有显著差异(图
4G
)。基于许多关于
M1/2
微胶质细胞的已发表的体内研究,我们注意到研究人员使用
Iba-1
,这不仅在微胶质细胞中表达,而且在巨噬细胞中也表达,作为微胶质标记。他们还使用
CD86
和
CD16
(由
Fcgr4
编码)标记
“M1”
微胶质细胞,使用
Arg1
和
CD206
(由
Mrc1
编码)标记“
M2”
微胶质细胞。然而,根据我们的
scRNA-seq
数据,
“M1”
微胶质细胞标记物(
Cd86
和
Fcgr4
)在微胶质细胞和巨噬细胞中均有表达,而
“M2”
微胶质细胞标记物(
Arg1
和
Mrc1
)主要在巨噬细胞中表达,表明渗透的巨噬细胞可能被错误地认定为经典定义的
“M2”
微胶质细胞(图
4H
)。因此,
M1/M2
的二分法可能无法识别不同的微胶质细胞亚群,并描绘它们在缺血后的功能。
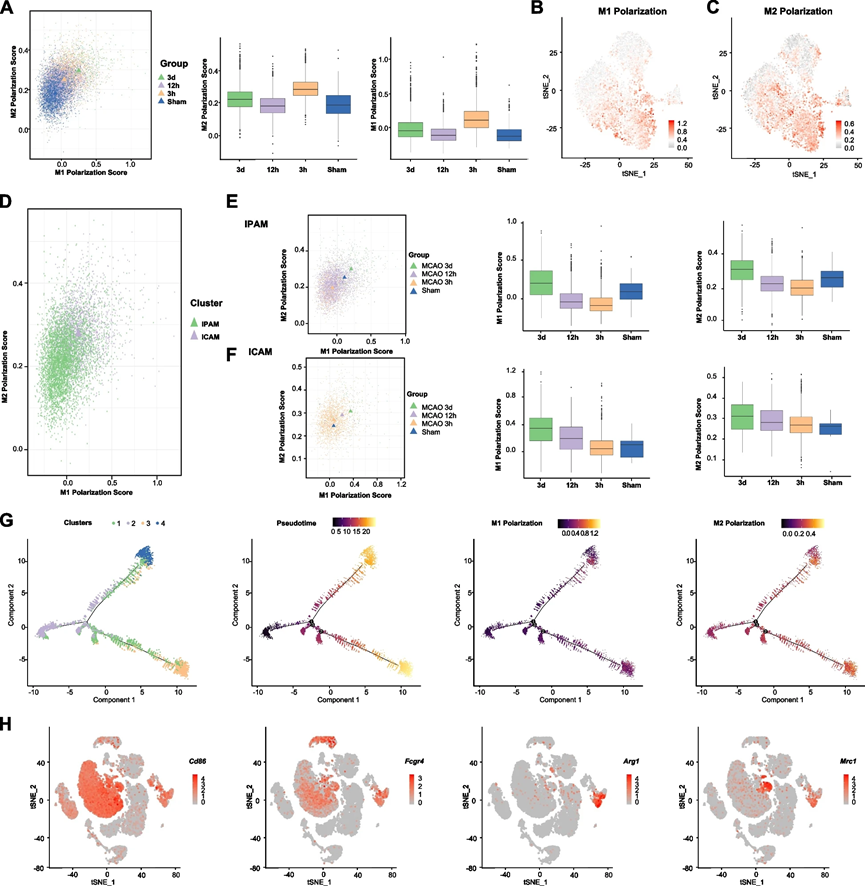
5
、关键
DAMP
和调节子驱动
ICAM
生成
我们假设区域微环境信号可能驱动微胶质细胞代谢和功能的转变,使其朝着
ICAM
和
IPAM
方向发展。通过使用
Ingenuity System Pathway Analysis
(
IPA
)软件(
QIAGEN
),我们发现脂多糖(
LPS
)是推动
ICAM
生成的最重要的潜在因素之一(图
5A
)。由于
LPS
是
TLR4
激活的经典配体,我们调查了在脑缺血后释放的
TLR4
内源性配体
HMGB1
的空间分布。在
MCAO
后的
12
小时内,在梗死核内受损神经元内明显出现
HMGB1
的胞质转位,这表明
HMGB1
,作为一种被广泛认识的损伤相关分子模式(
DAMP
),在梗死核释放并可能驱动
ICAM
的形成(图
5B
)。我们发现在
LPS
刺激后,大多数
ICAM
特异性标记基因在初级微胶质细胞中显著上调,而
IPAM
标志基因的表达变化较小(图
5C
,
D
)。为了调查垂死神经元释放的
DAMP
是否诱导
ICAM
的生成,我们还通过多次冻融小鼠神经细胞系
HT-22
制备了神经源性的
DAMP
,如先前描述的 。结果表明,处理
HT-22
源
DAMP
的初级微胶质细胞显著上调了
ICAM
标记基因,而
IPAM
标记没有显著差异。
除了细胞外的微环境信号外,我们还研究了哪些细胞内调控因子驱动
ICAM
的生成。使用单细胞调控网络推断和聚类(
SCENIC
)分析 来阐明缺血性卒中后微胶质细胞亚群形成的关键分子机制。通过全面考虑调控子活性评分(
RAS
)和调控子特异性评分(
RSS
),我们确定
FOSL1
和
BACH1
是最强大的
ICAM
特异性转录因子(
TFs
)之一(图
5E
,
F
)。研究表明,在
ST
中,
Fosl1
在急性缺血后在缺血区域上调和富集。此外,
BACH1
下游靶基因的富集分析(使用
Metascape
)发现它们与炎症显著相关(例如,“细胞迁移的正调控”,“
MAPK
级联的正调控
”
,
“TNF-α NF-κB
信号通路
”
,
“
细胞因子产生的调控
”
,
“MyD88
无依赖
TLR4
级联
”
,
“IL-17
信号通路
”
,
“
对氧化应激的应答
”
和
“
模式识别受体信号通路
”
),这与
ICAM
的促炎作用一致。为了验证
BACH1
对
ICAM
的调控能力,我们转染了携带靶向
Bach1
的短发夹
RNA
(
shRNA
)的慢病毒的
BV2
细胞。
Bach1-knockdown
在
LPS
刺激后下调了
ICAM
特异性标记基因(
Edn1
,
Plau
)的表达水平,表明
BACH1
有潜力将稳态微胶质细胞转化为
ICAM
。此外,
LPS
刺激后,经典的促炎因子,包括
Tnf
,
Il1a
,
Il6
和
Il1b
,在
Bach1-knockdown
细胞中也下调了(图
5G-J
)。
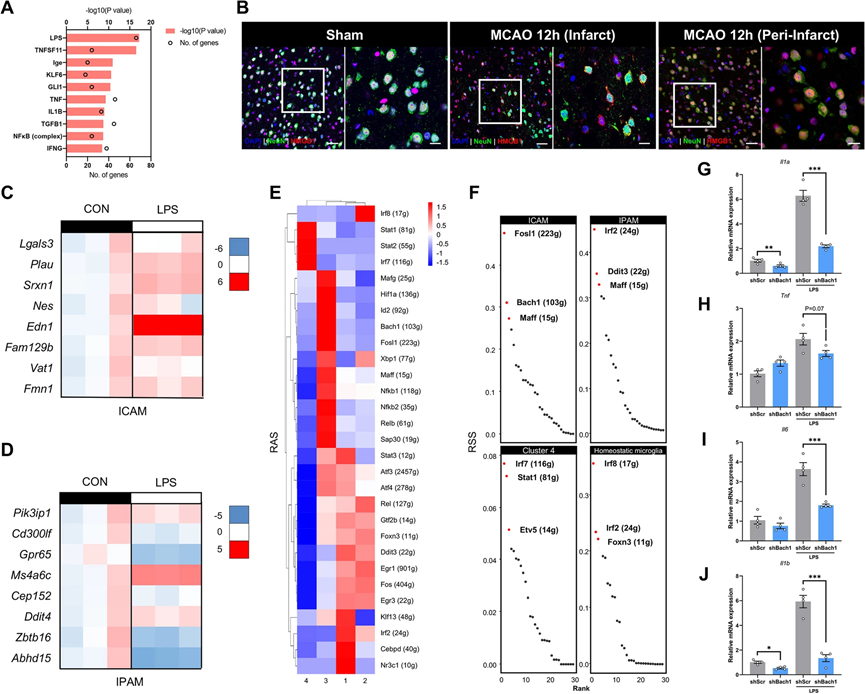
6
、糖皮质激素触发
IPAM
形成
为了识别
IPAM
的关键上游调控因子,使用了前
200
个
IPAM
标记基因进行
IPA
。令人惊讶的是,地塞米松(
DEX
)是诱导
IPAM
最强大的刺激因素(图
6A
)。为了探索中风后糖皮质激素浓度的变化,我们进一步利用高通量靶向代谢组学来筛选在缺血后显示变化的类固醇。根据定量数据,皮质酮和
11-
去氧皮质酮的水平在
MCAO
后在缺血核心和梗死周围区域均显著增加(图
6B
)。为了验证,我们使用
DEX
刺激初级微胶质细胞,强烈诱导了
IPAM
的生成,并显著增强了
IPAM
特异性标记基因的表达(图
6C
)。此外,小鼠主要的肾上腺皮质类固醇皮质酮也显著上调了初级微胶质细胞中
IPAM
特异性标记基因的表达,表明糖皮质激素具有推动
IPAM
生成的强大能力(图
6D
)。
为了在小鼠中验证这些发现,对
MCAO
小鼠进行了皮质酮或糖皮质激素受体拮抗剂(
RU486
)的处理。令人惊讶的是,接受皮质酮处理的
MCAO
小鼠显示出相对较轻的神经功能缺陷(图
6E-I
)和减少的神经元凋亡(图
6J
,
K
)。然而,在小鼠中
RU486
的投与导致更严重的神经功能缺陷(图
6E-I
)和加剧的神经元死亡(图
6J
,
K
)。
为了在不同实验组中研究
IPAM
的变化,我们采用了免疫染色并测量了微胶质细胞中
PIK3IP1
的表达。与对照组相比,
PIK3IP1
的平均荧光强度在皮质酮处理组中显著更高,而在
RU486
处理组中相对较低(图
6L
,
M
)。因此,这些体外和体内的发现验证了糖皮质激素在推动
IPAM
生成以及在缺血性卒中的急性阶段中
IPAM
的神经保护性质中的决定性作用。
实验方法:
慢病毒转导、梗塞体积测量、神经行为测试、
t-SNE
降维、
scRNA-seq
、
GSVA
、伪时间分析、轨迹推断、
SCENIC
、
TUNEL
染色、空间转录组测序、实时定量
PCR
、
Western blot
、
RNAscope
、靶向代谢组学。
参考文献:
Li, H., Liu, P., Zhang, B. et al. Acute ischemia induces spatially and transcriptionally distinct microglial subclusters. Genome Med 15, 109 (2023).

