


【珍藏版】伯晓晨/陈河兵/郑晓飞全面总结基于DNA序列和表观遗传信息预测染色质相互作用和高级结构算法

责编丨兮
高通量染色体构象捕获技术
(high-throughput chromosome conformation capture,
Hi-C
)和
配对末端标签测序分析染色质相互作用技术
(chromatin interaction analysis by paired-end tag sequencing,
ChIA-PET
)的发展,极大地推动了染色质高级结构的研究。然而,高分辨率的Hi-C和ChIA-PET数据获取成本仍较高,目前可用的高分辨率数据十分有限。研究表明,DNA序列和基因组表观遗传修饰信息可用于染色质相互作用和高级结构的预测,目前已有多种相关算法被开发出来,但这些方法在生物医学领域还没有得到广泛应用。
近日,军事医学研究院
伯晓晨
和
郑晓飞
团队在
Briefings in Bioinformatics
杂志上发表方法学综述:
Computational methods for the prediction of chromatin interaction and organization using sequence and epigenomic profiles
,
分类总结了基于DNA序列和基因组表观修饰信息预测染色质相互作用和高级结构的48种计算方法,评估了不同方法的性能,并分析了相关的应用场景,为生物学家使用相关计算工具提供了系统地参考
。


基因组三维结构是DNA复制、表达调控、变异和进化的基础,阐明基因组功能是理解遗传物质如何决定细胞命运的关键。研究基因组三维结构,了解基因组的生物学功能,需要关注两个方面:一方面是
染色质相互作用
(chromatin interaction),即染色质上转录调控元件间的相互作用;另一方面是
染色质高级结构
(chromatin organization),约2米长的DNA在6-10微米的细胞核中复杂折叠,折叠形成的染色质高级结构决定着基因位点之间的相互作用频率。
染色体构象捕获技术的发展与应用,加深了我们对染色质多层级高级结构的认识(图1)。细胞核内每条染色体都占据着一个独特且不重复的区域,称为染色体疆域(chromosome territory, CT),染色体疆域中包括与常染色质和异染色质分别相关的A、B染色质区室(compartment)。染色质区室由拓扑相关结构域(topologically associated domain,TADs)组成,并可进一步细分为染色质环(chromatin loop),如调控基因表达的增强子-启动子环[1]。环挤压(loop extrusion,LE)模型认为TAD的形成是由主动挤压机制介导的,环状挤压因子-黏连蛋白(cohesin)沿着染色质移动,当它们遇到另一个挤压因子或两个相反方向的边界元素-CTCF(CCCTC binding factor,CTCF)时,将其挤压成环[2]。
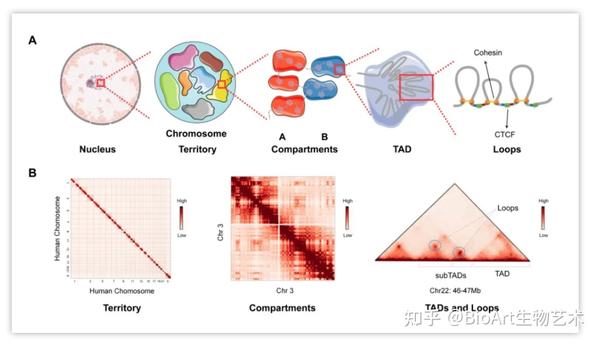

图1:
多层级染色质高级结构
。(A)多层级染色质高级结构。从左到右为细胞核,染色质疆域,染色质区室,拓扑关联结构域和染色质环。(B)Hi-C相互作用矩阵。染色体疆域;3号染色体内的染色质区室;22号染色体上的TAD,subTAD和loop结构,loop表示为点状信号。
染色质相互作用预测方法
我们总结了33种预测染色质相互作用的方法,其中包括12种非监督学习方法和21种有监督学习方法(图2,表1),预测的染色质相互作用主要可分为三类:增强子-启动子相互作用(enhancer-promoter interaction,EPI)、增强子的靶标基因(enhancer-target genes,ETGs)和3D染色质相互作用(3D interaction)。
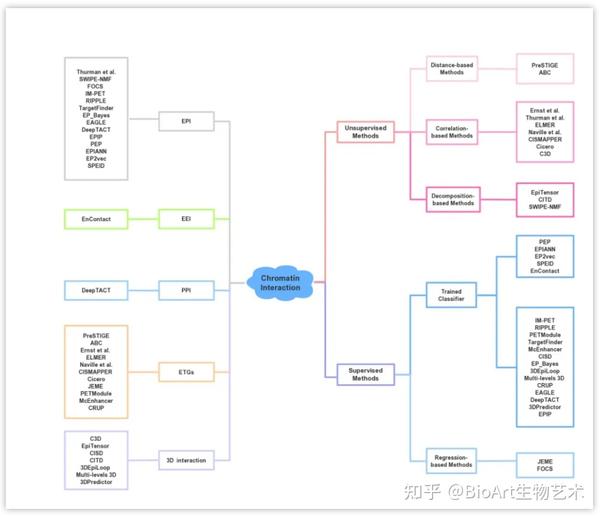

图2: 染色质相互作用预测方法 。左侧按照预测结果分类,右侧按照工具类型分类。
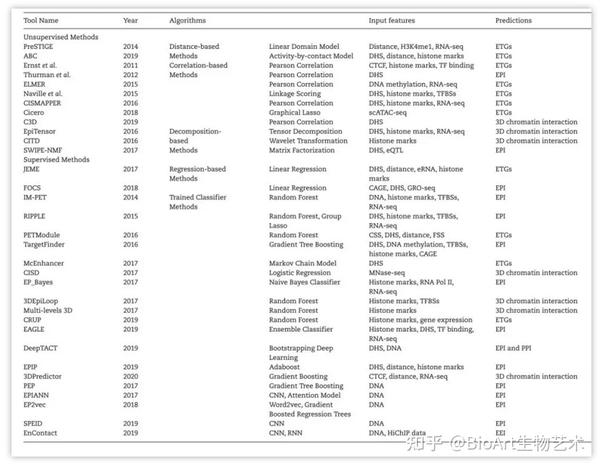

表1:染色质相互作用预测方法
非监督学习方法
非监督机器学习方法通过分析染色质调控元件间的距离及表观修饰间的相关关系,揭示染色质相互作用的自然发生模式,来预测染色质相互作用[3]。根据预测策略不同,这些非监督学习方法又可以分为基于距离的方法、基于相关性的方法以及基于分解的方法。
基于距离的方法
。通过计算染色质调控元件之间的线性距离,将增强子分配给距离其最近的基因,可以有效地预测EPI。基于距离的预测方法原则简单,该方法在许多研究中作为基线方法来衡量其他方法的预测性能。PreSTIGE 方法(
http://
prestige.case.edu/
)[4]是一种用于预测ETGs的多线性模型,它通过整合基因表达RNA-seq数据和组蛋白修饰H3K4me1的ChIP-seq数据,将CTCF结合位点作为绝缘子,对细胞特异性增强子与其靶基因进行关联[5]。由于PreSTIGE方法仅基于距离和简单的相关策略,其预测准确性较低[5]。ABC方法[6]通过距离表征增强子-启动子对相互作用频率,并结合增强子活性信号预测ETGs。
基于相关性的方法
。在基于距离方法的基础上,基于相关性的方法结合了组蛋白修饰,增强子和启动子的脱氧核糖核酸酶I超敏信号(DNase I hypersensitive signals,DHSs)及基因转录水平等特征的相关性,提升了预测的准确性。Ernst团队[7]利用转录因子(transcription factors,TFs)表达与转录因子基序(motif)富集的相关性预测细胞类型特异性靶基因的调控元件。Thurman团队[8]通过分析调控元件的DHSs信号水平预测EPI。Cicero方法[9]基于单细胞ATAC-seq数据预测ETGs。C3D方法[10]根据染色质开放区域之间的相关性预测EPI。Naville团队[11]基于染色质可及性、组蛋白修饰和TFBSs之间的相关性预测ETGs。ELMER方法[12]将增强子甲基化程度与邻近基因的表达关联识别ETGs。在ELMER的基础上,Silva团队开发了ELMER v2[35],该版本提供了新的监督预测模型和网页服务器。CISMAPPER方法[13]基于组蛋白标记与基因表达的相关性预测EPI。
基于分解的方法
。该类方法通过分解表观遗传修饰的高维信号,提取可用于预测染色质相互作用的潜在特征,并根据潜在特征之间的关系来识别染色质相互作用。EpiTensor方法[14]从一维表观修饰信号构建3D染色质相互作用,它使用张量表示多种细胞类型中的高维表观遗传修饰数据,并将张量分解为细胞类型,表观遗传修饰信号和基因组位点等子向量。基于张量分解的子向量之间的关联,Epitensor方法可以识别染色质相互作用中的热点启动子和增强子,它们位于基因组具有显著转录活性和转录因子结合位点富集的区域[14]。CITD方法[15]基于小波分解和组蛋白修饰信号重建整合一维组蛋白修饰数据预测染色质的相互作用。SWIPE-NMF方法[16]构建了矩阵分解框架,整合了不同类型表观遗传数据,并重建了127个人类细胞系的增强子-启动子网络。
监督学习方法
监督学习方法主要包括随机森林、神经网络、决策树、Logistic回归和线性回归分析等。根据模型中应用的算法的不同,监督方法可分为基于回归的方法和训练分类器。
基于回归的方法
。基于回归的方法将增强子和启动子特征或基因表达相关联,识别增强子与启动子之间的相互作用或与靶基因间的调控关系,如JEME和FOCS方法[17, 18]。JEME方法[17]考虑了多个增强子联合作用的情况并整合了总体特征和样本特异性信息,可用于预测基因调控网络。FOCS方法[18]是一个统计框架,该方法使用eRNA作为增强子活性的标记,根据染色质表观修饰信息来判断与转录活性相关的增强子-启动子相互作用。
训练分类器
。这类方法以Hi-C实验和ChIA-PET实验识别的增强子-启动子相互作用作为金标集,通过学习这些相互作用增强子和启动子的序列及表观修饰特征,可以训练分类器,预测给定启动子-增强子对是否存在相互作用关系。IM-PET方法[19]通过整合增强子活性、启动子活性、TFs-启动子相关性,相互作用增强子-启动子的进化保守性以及增强子与目标启动子之间的距离约束等特征来识别EPI。
PETModule方法[20]是一种基于motif的ETGs预测方法,它使用了与IM-PET相似的特性。RIPPLE方法和TargetFinder方法[21, 22]根据染色质开放状态、基因表达水平、TFs、结构蛋白和组蛋白修饰水平等多种特征来识别细胞类型特异的EPI。EAGLE方法[23]可以应用于不同物种和细胞类型的数据,使用少量的表观修饰特征就可具备较高的预测准确性。EPIP方法[24]将多种表观修饰特征分为11个集合,在缺少某些数据的情况下仍然可以预测EPI。EP_Bayes方法[25]使用RNA聚合酶II的ChIP-seq数据作为输入,不适用于内含子增强子相互作用的预测。使用DHSs作为输入特征,McEhancer,CISD和DeepTACT方法[26-28]可用于预测多种类型的染色质相互作用。其中,McEhancer方法[26]可以预测ETGs。CISD方法[29]根据核小体排列模式特征识别全基因组染色质的相互作用位点。DeepTACT方法采用自举检验(Bootstrapping)方法整合序列和染色质可及性的数据,预测EPI和启动子-启动子相互作用(promoter-promoter,PPI)。另外,还有一些工具可以用于预测3D染色质相互作用。3DEpiLoop方法[30]可以用一维表观基因组和转录因子结合位点预测TAD中的高分辨率(1kb)3D染色质相互作用。Bkhetan方法和Plewczynski方法[31]提出了一种随机森林分类器,可以通过表观基因组图谱预测3D染色质相互作用。3Dpredictor方法[32]基于CTCF结合信号和基因表达数据可以预测3D染色质相互作用。
还有一些工具可以仅基于DNA序列信息作为输入即可预测调控元件之间的相互作用。PEP方法[33]整合了PEP-Motif和PEP-Word两个模型,通过从特定细胞类型的增强子和启动子位置提取序列特征来预测EPI。SPEID方法[34]应用深度神经网络(deep neural networks,DNN),根据增强子和启动子区域的序列特征来预测EPI。由于缺乏基于序列的通用EPI预测机制,SPEID方法只能在训练细胞系中有效地预测EPI[34]。EPIANN方法[35]是一种基于注意的神经网络模型(attention-based neural network model),该方法更专注于EPI的特征,准确地预测EPI。EP2vec方法[36]采用自然语言处理的非监督深度学习方法,将增强子和启动子序列转化为序列嵌入特征,并采用监督分类器对EPI进行预测。EnContact方法[37]是一种以DNA序列作为输入预测增强子-增强子相互作用(enhancer–enhancer interaction,EEI)的深度学习模型。
染色质高级结构预测方法
目前已有很多机器学习方法和多聚物理模型应用于染色质高级结构的预测(图3, 表2)。
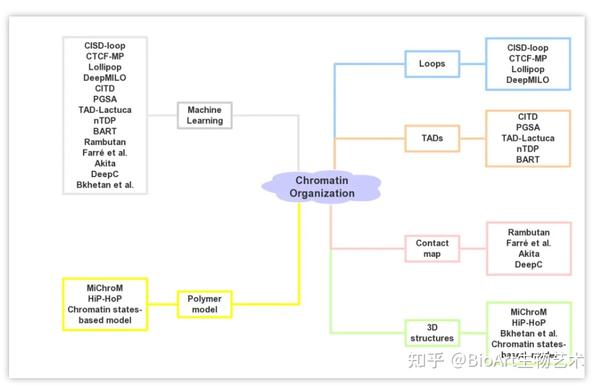

图3:
染色质高级结构预测方法
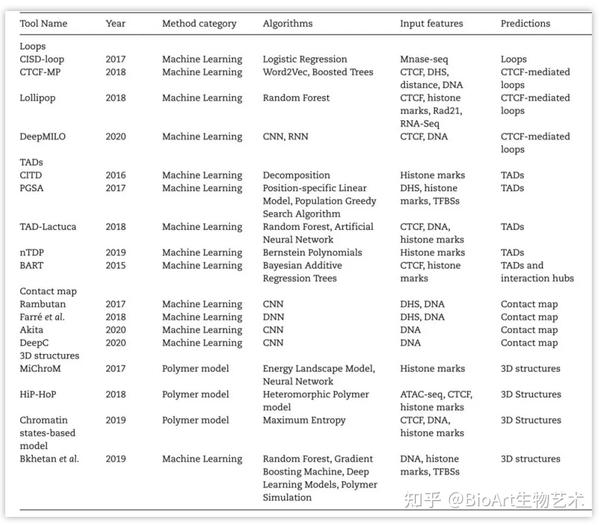

表2: 染色质高级结构预测方法
Loop预测
。基于染色质相互作用可以改变核小体侧翼排列模式特征的假设,CISD-loop方法[29]整合了微球菌核酸酶敏感位点测序(micrococcal nuclease sensitive sites sequencing,MNase-seq)数据和低分辨率Hi-C数据,可以在多种人细胞系中准确地预测染色质环。Word2vec是一种常用的自然语言处理的神经网络模型,CTCF-MP方法[38]通过Word2vec和提升树提取CTCF的ChIP-seq和DNase-seq数据特征,预测CTCF介导的染色质环。Lollipop方法[39]是一种随机森林分类器,它根据DNA序列和表观修饰信息特征来区分两个染色质区域是否形成CTCF介导的染色质环。DeepMILO方法[40]提供了用于预测CTCF介导染色质环的深度学习网络,并可以通过DNA序列数据预测非编码区突变对染色质环形成的影响。
TAD预测
。CITD方法[15]基于多种组蛋白修饰在染色质相互作用位点的相关性,根据染色质相互作用频率随距离而下降的规律,整合了组蛋白修饰数据预测染色质的相互作用频率、TAD边界及TAD的转录活性。PGSA方法[41]通过基因组上与TAD形成相关的基因组元件,如CTCF、ZNF143和YY1等来预测TAD边界。同样,TAD-Lactuca方法[42]根据DNA序列和8个组蛋白标记预测基因组某一位点是否位于TAD边界。nTDP方法[43]采用半监督非参数的模型框架,使用组蛋白修饰H3K36me3、H3K4me1、H3K4me3和H3K9me3数据进行TAD预测。BART方法[44]可用于预测细胞类型特异性组蛋白修饰信息的染色质相互作用中心和TAD边界。
相互作用图谱预测
。Hi-C数据得到的染色质相互作用图谱(contact map)可用于多层级染色质高级结构的识别。Rumbutan方法[45]是一种基于深度卷积神经网络的计算工具,可用于没有Hi-C数据的情况下,使用DNA序列和DHSs作为输入预测Hi-C相互作用图谱。Farré团队[46]基于DNA序列训练密集神经网络,进而预测染色体内的相互作用,他们强调染色质上下游序列及活化状态在邻域构象形成中具有重要作用。Akita方法[47]是一个卷积神经网络(convolutional neural network,CNN),该方法仅需DNA序列作为输入就能预测染色质相互作用图谱。同样,基于深度神经网络(Deep Neural Networks,DNN)的DeepC方法[48]也可通过DNA序列信息预测染色质相互作用图谱,DeepC可以预测更精细的域间相互作用。
染色质结构仿真建模
。MiChroM方法[49]是一种能量景观模型,基于组蛋白修饰模式与染色质高级结构间的关系预测染色质高级结构。HiP-HoP方法[50]是一种基于转录因子和环挤出模型的异质性聚合模型,该方法通过DNA可及性、H3K27ac和CTCF/Rad21信号来预测染色质的多聚结构。Qi和Zhang团队[51]提出了一种可转移的聚合模型,用于模拟5kb分辨率的染色质3D结构,该模型以基因组位置、表观修饰特征和CTCF方向作为输入。Bkhetan团队[52]改进了3DEpiLoop方法,并结合Spring模型,构建了基于分子力学的染色质3D结构可视化方法。
预测方法的性能比较
由于不同方法使用的预测策略及输入数据不同,难以找到一个金标准对所有工具进行系统性比较。常用的计算模型评价指标包括F1值、接受者操作特征曲线下面积(area under the receiver operating characteristic curve,AUROC)和召回率和正确率组成曲线下面积(area under the precision-recall curve,AUPR)。F1值是精密度(precision)和召回率(recall)的调和均值。AUROC和AUPR不依赖于特定的分类器阈值,AUPR相较于AUROC对不平衡的数据更加敏感。我们根据输入数据和算法分类总结了不同类型方法的性能和优缺点。
不同输入数据计算方法的性能比较
计算工具对输入数据的要求不尽相同,TargetFinder和RIPPLE需要多个表观修饰信息判别增强子-启动子对是否相互作用,而EP2vec、PEP和SPEID等工具则仅以DNA序列作为输入就可以预测EPI[21, 22, 33, 34, 36]。Zeng团队基于TargetFinder的(E/P/W)训练数据集,评估了EP2vec,TargetFinder和SPEID的性能,发现与TargetFinder相比,EP2vec的F1值略高,而AUROC和AUPR较低,在10倍交叉验证测试中,这两种方法均优于SPEID(表3)[36]。Yang团队使用TargetFinder的(EE/P)数据集验证表明,PEP和TargetFinder的性能相近,且两种方法均优于RIPPLE,具有更高的F1值,AUROC和AUPR(表3)[33]。这些结果表明,仅使用序列数据足以预测染色质的相互作用,结合表观遗传信息可以进一步提高预测的准确性。
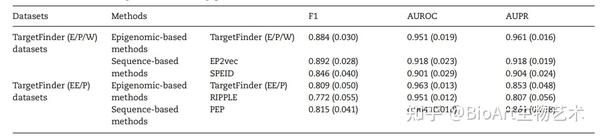

表3: 基于DNA序列方法与基于表观遗传信息工具性能的比较
SPEID方法[34]通过减少预测特征,识别预测染色质相互作用的必需特征。TargetFinder方法与SPEID方法不同,TargetFinder在训练过程中,通过添加预测特征,判断模型的预测性能,导致TargetFinder虽然有较好的预测表现,但要达到相应的预测准确性必须具有很多种输入数据,考虑到细胞系的表观遗传数据往往有限,这可能会限制工具的使用[22]。我们用TargetFinder的六个细胞系的训练数据集(E/P/W)检验了TargetFinder输入特征的冗余性。通过对输入特征的重要性进行排序,并根据它们的预测重要性递归地消除基因组特征,进行10倍交叉验证,输出模型的F1值(图4)。结果表明TargetFinder使用的输入特征具有一定冗余性,尤其是GM12878和K562细胞系。当具有多种表观遗传信号时,TargetFinder具有更好的性能,而当只有几种核心特征可用时,TargetFinder也可以实现较好的性能。Moore团队使用来自101个表观基因组数据集的303个特征和四个核心表观基因组特征:DNase-seq、H3K4me3、H3K27ac和CTCF数据以及距离特征分别对TargetFinder的预测性能进行了检测。他们发现使用四个核心特征用于预测时,平均AUPR降低23%,降低后的AUPR值仍然比基于距离的预测方法效果[53]。
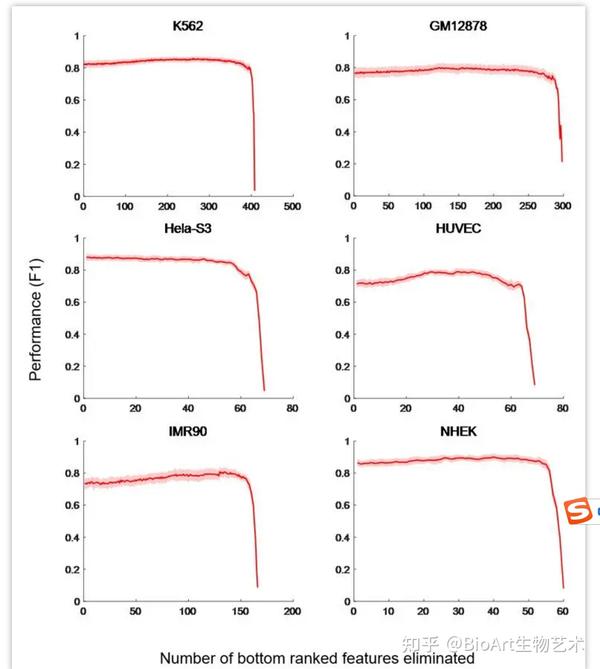

图4: 预测特征减少的TargetFinder性能
监督和非监督方法的性能比较
Moore团队开发了候选增强子-基因相互作用(Benchmark of candidate Enhancer–Gene Interactions,BENGI)评价基准,并用该基准比较了非监督和监督计算方法的性能(图5A)[53]。Moore团队比较了基于距离的方法,三种基于相关性的方法和两种监督学习模型,其中TargetFinder的预测效果最好,但在跨细胞系测试时,它的性能并不优于基于距离的方法[53]。结果还显示基于DNase-DNase相关性和DNase-基因表达相关性的方法并不优于基于距离的方法,这可能是细胞系之间的相关性不稳定导致的[53]。此外,当使用GM12878的六个BENGI数据集进行交叉验证时,TargetFinder的性能明显优于PEP-Motif。基于距离的方法依赖于精确的调控元件识别,而忽略了增强子跳过附近基因作用于更远端靶基因的可能性[53]。理论上讲,基于相关性的方法应该优于基于距离的方法,而某些相关性可能在细胞系之间不稳定,导致了其次优性能。
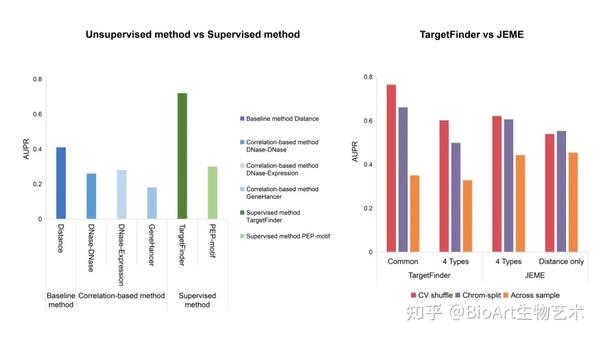

图5:
非监督和监督方法的性能
。(A)使用BENGI数据集对基于距离,基于相关性和监督学习方法的性能比较[53]。(B)基于回归的模型和训练分类器方法的性能比较[53]。
此外,一些监督方法由于使用了有问题的交叉验证策略而产生了过度拟合。使用TargetFinder使用的6个(E/P/W)数据集,Cao团队发现window区(增强子与启动子之间的染色质区域)特征之间的高度相似性,训练集和测试集样本的相似性会夸大交叉验证结果,这也解释了为什么TargetFinder跨细胞系使用时预测效果不理想[54]。为了打破训练和测试集中样本之间的依赖关系,Cao团队引入了染色体分裂策略,将同一染色体上的所有样本分配给训练或测试集[54]。在JEME方法使用的数据集上,通过交叉验证对TargetFinder和JEME进行比较,结果显示染色体分裂策略的AUPR远低于随机分配的AUPR值(图5B)[54]。此外,TargetFinder使用K562和GM12878共有的特征比使用四种表观特征(H3K4me1、H3K27ac和H3K27me3的DNase-seq和ChIP-seq)表现得更好,而JEME使用四种特征的性能与仅使用距离特征相似(图5B)[54]。
DeepMILO方法预测染色质环的性能优于CTCF-MP方法,表明相较于常规的机器学习模型,深度学习模型可以更好地学习复杂的序列特征,以有效地预测突变对loop的影响[40]。深度学习是机器学习研究中最活跃的领域之一,最近已经应用于基因组学相关的各项研究中,如功能基因组注释和基因表达预测等[55]。深度学习可以通过训练具有多个层次的复杂网络,自动从大量的高维数据中提取复杂而有意义的特征,集成不同类型的输入数据。深度学习不仅提高了预测性能和准确性,通过其框架中每一层代表的含义还能够对基因组三维结构建立的机制进行探索。
一般来说,监督学习方法受到训练标签不确定性的限制,监督学习的标签需要根据生物学专业知识进行定义,定义的不准确可能会影响预测结果。相比之下,非监督学习的方法不使用先验知识,可以发现新的染色质相互作用模式,如PPI和EEI等。然而,通过非监督学习发现的新相互作用模式的验证又是新的挑战。目前已有半监督方法来预测染色质相互作用和高级结构,它们通过非监督的方法在DNA序列及基因组表观遗传信息中提取染色质相互作用或高级结构相关特征,并将初始特征和提取特征的模型用于监督模型的训练并进行预测,该方法较监督学习模型可能具有更好的性能[40, 43]。
使用指导及方法推荐
使用计算方法对染色质相互作用和高级结构进行预测包括三个步骤:(1)从数据库或实验获取输入数据;(2)选择计算方法输入数据;(3)输出表示染色质相互作用或高级染色质结构的结果。在选择计算工具时,可以基于预测目标或已有数据进行选择(图6)。
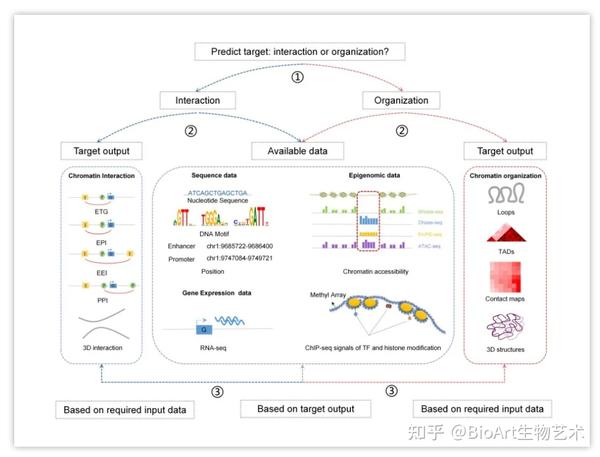

图6: 计算方法的选择
要选择合适的方法,首先应该确定预测目标(染色质相互作用或高级结构),然后用户可以根据现有数据或目标输出进一步选择方法。如果用户已经获得了实验数据,则可以优先考虑根据输入数据进行选择。如果优先考虑输出结果,则用户可以根据方法的输出结果进行选择,再准备该方法所需的输入数据。
表4中列出了几种输入数据要求较少,使用方便的工具供大家选择。PreSTIGE是一种基于Web的工具,使用H3K4me1的ChIP-seq数据和RNA-seq数据预测ETGs[5]。Cicero使用单细胞ATAC-seq数据预测ETGs[9]。C3D和McEnhancer可根据DNase-seq数据预测EPI和3D染色质相互作用[10, 26]。ELMER用甲基化修饰信息和RNA-seq数据预测ETGs[12]。EPIANN和EP2vec仅基于DNA序列数据识别EPI[35, 36]。染色质高级结构识别的工具中,DeepMILO可以根据DNA序列和CTCF信号预测CTCF介导的染色质环。TAD-Lactuca使用组蛋白修饰信息预测TAD边界。Akita仅需DNA序列数据预测染色质相互作用图谱[42]。HiP-HoP使用ATAC-seq或DNase-seq,H3K27ac,CTCF和Rad21数据,预测染色质3D多聚模型[50]。


表4: 方法推荐
生物医药研究应用
大数据时代,计算工具的应用在医药领域研究中必不可少。我们总结了相关计算方法在生物领域的应用实例,主要包括三个方面:基因突变对基因表达的影响,基因调控和疾病发生进展机制的研究。
基因突变对基因表达的影响
。全基因组关联研究(genome-wide association study,GWAS)中发现多数非编码单核苷酸多态性(single nucleotide polymorphisms,SNP)通常位于细胞系特异性增强子中,并与多种常见疾病的风险相关[56, 57]。PreSTIGE方法的分析结果表明多个增强子突变共同促进了其基因靶标的表达变化,并证明了GWAS鉴定的SNP具有改变某些特定性状的风险,非编码区突变赋予了某些性状的突变易感性[5]。ABC方法的分析结果解释了影响人类性状的非编码性遗传突变的功能[6]。Ernst团队开发的染色质相互作用预测工具可以将疾病相关的性状与疾病相关突变联系起来[7]。CTCF-MP方法可以解释突变引起的序列变化,并预测这些突变对染色质环的影响[38]。DeepMILO方法可以预测GWAS鉴定的样本突变对CTCF介导的染色质环的影响[40]。基于C3D,Johnstonet团队验证了染色质结构突变对基因组3D结构和转录的影响[10]。PRISMR方法可以识别由疾病相关的染色质结构变异引起的染色质相互作用,这些相互作用的改变可能会使TAD分布改变,从而导致基因的表达异常[58]。DeepC方法可以预测大规模染色质结构变异和单个碱基变异对染色质高级结构的影响[48]。
基因调控机制研究
。上述计算方法还被用于研究基因表达调控和染色质高级结构的形成机制。Cicero方法可用于研究染色质可及性变化对邻近基因表达的影响并分析基因组范围内的顺式元件调控网络[9]。DeepTACT方法可用于识别热点启动子,这些启动子在多种细胞系中活跃并多调控管家基因的表达,DeepTACT的分析结果表明,IFNA2可能是重要的自身免疫靶标基因[28]。同样,EnContact方法可用于识别热点增强子。Moquin团队扩展了TargetFinder的应用,将预测相互作用的一个位点设在人类基因组中,另一个位点设在Epstein–Barr(EB)病毒的基因组中,以研究EB病毒是否与人类基因组具有相似的转录调控机制[59]。Schreiber团队使用Rambutan方法为53个人组织生成了染色质相互作用图谱,发现具有相似功能的细胞类型具有相似的染色质高级结构,而癌细胞中则没有这个规律[45]。
疾病发生发展机制
。上述计算方法还应用到了疾病相关机制的研究中。ELMER方法是一种研究癌症相关TFs及其靶基因之间顺式调控关系的算法,可用于研究TCGA数据库中癌症特异的增强子和启动子[12]。Ravi团队用ELMER v.2进行了甲状腺癌的mRNA表达和DNA甲基化分析,结果表明,DNA异常甲基化会影响基因表达,并可能促进肿瘤发生[13]。此外,Naville团队使用ChIP-seq数据预测了MCF7细胞系中的染色质的相互作用[25],根据此结果Dzida等验证了这些细胞对雌二醇的早期反应[11]。Sin-Chan团队在多层玫瑰花结的胚胎肿瘤中使用C3D分析了其远距染色质相互作用,发现了潜在的治疗位点[60]。
此外,这些方法还提供了可用于生物医学问题研究的预测结果。例如,PreSTIGE方法提供了2695个ENCODE注释的lncRNA组织特的增强子,以及13个细胞系的染色质相互作用数据库[5]。这些预测目前已被应用到头颈部鳞状细胞癌eRNA-靶基因的识别[61],确定影响体重指数的染色质相互作用[62],预测感官上皮细细胞发育和成熟相关的调控机制[63],以及分析调控区域中SNP与相关疾病的关联[64]。
JEME方法重建了935个人的原代细胞,组织和细胞系样本的增强子-启动子网络,所得的预测已用于研究与哮喘[65],骨密度[66],注意力缺陷/超敏性障碍[67],血清中的前列腺特异性抗原[68],精神分裂症[69],骨关节炎[70]和免疫应答[71]等相关的生物医学问题的研究。上述方法的预测结果被SEdb和OncoBase等数据库收录,可供生物研究人员使用研究相关疾病的调控机制[72, 73]。
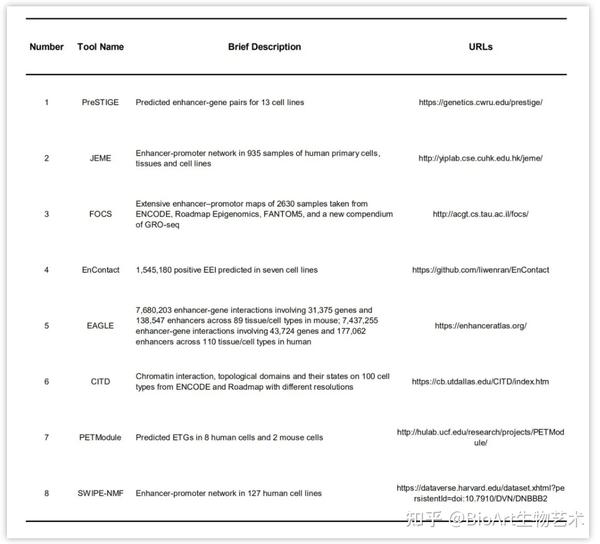

表5: 可用预测结果数据库
讨论与展望
染色质相互作用和高级结构的精准预测对于探究基因表达调控机制、细胞分化和疾病发生机制至关重要。基于序列和表观修饰数据的计算方法极大地提高了我们对染色质3D结构及其转录调控作用的理解。我们描述了现有的基于DNA序列和表观基因组数据的染色质相互作用和高级结构的预测方法,该工作指导了如何根据现有数据或目标输出选择合适的计算方法,并比较了这些方法的性能,总结了各种方法的优缺点。通过介绍不同方法在生物医学领域的应用实例,进一步帮助了研究人员选择合适的研究方法。
染色质相互作用及高级结构预测面临的挑战
。在染色质相互作用和高级结构预测中仍存在很多挑战。首先,高分辨率Hi-C数据仍然有限,数据分辨率限制了对更精细染色质高级结构的探索。目前已经开发了多种Hi-C分辨率提升算法,如DeepHiC[74],HIFI[75],Boost-HiC[76],deDoc[77],hicGAN[78],HiCNN[79]和HiCPlus[80]等,可以通过低分辨率数据得到高分辨率数据。其次,染色质高级结构的计算方法仍然存在局限性。基于距离的方法忽略了远端调节相互作用,以及多个增强子靶向同一启动子的情况。用于识别增强子-启动子对的相关性统计方法的高灵敏度使预测结果具有相对较高的假阳性。第三,目前仍然缺乏探索单个细胞中精确识别染色质高级结构的方法。
染色体某些结构在多个细胞系间表现出较高的异质性。大多数现有方法使用总体平均数据,并专注于拟合单一染色质结构,构建模型时忽略了单个细胞变异和细胞间染色质结构的变异性。因此,单细胞Hi-C技术和基于表观基因组数据的计算方法应被更加广泛的使用。第四,目前检测染色质相互作用的实验技术仍不够理想,实验噪声对预测结果的置信度影响较大。使用序列和表观基因组数据的计算方法基于生物假设来预测染色质相互作用,如关于特征性核小体排列,染色质状态的相关性,序列motif特征和进化保守策略,这些生物学假设为染色体构象捕获技术检测的染色质高级结构进行了补充。第五,Hi-C实验的分辨率受到限制性核酸内切酶的限制。近期开发的多种Hi-C衍生技术突破了该限制,例如Micro-C技术通过MNase将染色质片段化来克服限制性核酸内切酶分辨率的限制[81]。相对于Hi-C,Micro-C表现出更好的信噪比,且可以在哺乳动物细胞中捕获更精细的染色质的高级结构[81]。第六,当前计算方法的性能仍不够理想。用一个细胞系的数据训练好模型后,在其他细胞系中进行预测的效果往往较差。目前一些工具通过整合多种实验方法预测染色质的相互作用和高级结构,提高了预测性能,如BART方法整合了Hi-C和表观遗传特征[44],GEM-FISH方法整合了荧光原位杂交和Hi-C数据进行染色质高级结构的预测[82]。
关于改善用户体验的建议
。尽管目前已有多种预测染色质的相互作用和高级结构的计算方法,但这些方法在生物医学研究中的应用仍不广泛。由于某些计算方法的黑盒性质,它们与生物学功能的关联难以清楚阐明,且方法中包含的复杂算法对于缺乏计算知识的生物学家来说难以理解。这些方法采用了不同的软件和编程语言,包括python,java,C ++和R等,且一些方法提供的使用说明不够清晰,很多方法的计算结果难以重现。因此,在相关计算工具开发的时候,开发人员除了关注工具性能的同时,还应更加关注方法的用户体验,这将有助于促进这些方法的推广使用。基于网页的集成原始数据处理和分析工具,可以使用户通过简单的操作上传输入数据并获取输出文件,是最便于使用的工具形式。开发人员还应提供更详细的使用说明,包含原始数据的处理过程及运行环境、版本等信息,并提供简单的示例数据帮助用户尽快熟悉工具的使用方法。此外,可以提供有关运行进度、摘要报告和支持信息的反馈,使计算过程更加可视化。
随着基因组、转录组和表观遗传修饰数据的积累,以及数据处理能力的不断提高,用于预测染色质相互作用和高级结构的计算方法不断发展,极大地促进了染色质相互作用和高级结构的研究,为基因表达调控机制、细胞分化和疾病发生模式提供了新的生物学研究线索。比如,近期Kloetgen团队通过分析Hi-C,RNA-seq和CTCF ChIP-seq数据,发现了染色质3D结构在人类急性白血病中的复杂性和动力学特征,并证明通过小分子药物靶向染色质高级结构可以抑制疾病相关基因的表达[83]。对染色质高级结构的理解将为疾病相关的染色质高级结构突变提供新的见解,并促进以其为靶点的疾病治疗。
对于染色质高级结构的研究,通过计算工具预测调控元件相互作用和染色质高级结构的特征至关重要。相信随着计算方法的广泛应用和新的计算工具的开发,染色质三维结构与基因调控和疾病发展的关系将得到更加充分地揭示。
军事医学研究院
陶欢
硕士研究生和助理研究员
李昊
博士为该论文的第一作者,军事医学研究院
伯晓晨
研究员、
陈河兵
副研究员和
郑晓飞
研究员为该论文的共同通讯作者。北京大学
李程
研究员为该文提出了宝贵的指导意见。
原文链接:
https:// academic.oup.com/bib/ar ticle-lookup/doi/10.1093/bib/bbaa405
参考文献
1. Dixon J, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu J, Ren B: Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485(7398):376-380.
2. Fudenberg G, Abdennur N, Imakaev M, Goloborodko A, Mirny LA: Emerging Evidence of Chromosome Folding by Loop Extrusion. Cold Spring Harb Symp Quant Biol 2017, 82:45-55.
3. Xu H, Zhang S, Yi X, Plewczynski D, Li MJ: Exploring 3D chromatin contacts in gene regulation: The evolution of approaches for the identification of functional enhancer-promoter interaction. Comput Struct Biotechnol J 2020, 18:558-570.
4. Goecks J, Nekrutenko A, Taylor J, The Galaxy T: Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biology 2010, 11(8):R86.
5. Corradin O, Saiakhova A, Akhtar-Zaidi B, Myeroff L, Willis J, Iari RC-S, Lupien M, Markowitz S, Scacheri PC: Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits. Genome Research 2014, 24(1):1-13.
6. Fulco CP, Nasser J, Jones TR, Munson G, Bergman DT, Subramanian V, Grossman SR, Anyoha R, Doughty BR, Patwardhan TA et al: Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat Genet 2019, 51(12):1664-1669.
7. Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M et al: Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011, 473(7345):43-49.
8. Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B et al: The accessible chromatin landscape of the human genome. Nature 2012, 489(7414):75-82.
9. Pliner HA, Packer JS, McFaline-Figueroa JL, Cusanovich DA, Daza RM, Aghamirzaie D, Srivatsan S, Qiu X, Jackson D, Minkina A et al: Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Mol Cell 2018, 71(5):858-871.e858.
10. Mehdi T, Bailey SD, Guilhamon P, Lupien M: C3D: a tool to predict 3D genomic interactions between cis-regulatory elements. Bioinformatics 2019, 35(5):877-879.
11. Naville M, Ishibashi M, Ferg M, Bengani H, Rinkwitz S, Krecsmarik M, Hawkins TA, Wilson SW, Manning E, Chilamakuri CS et al: Long-range evolutionary constraints reveal cis-regulatory interactions on the human X chromosome. Nat Commun 2015, 6:6904.
12. Yao L, Shen H, Laird PW, Farnham PJ, Berman BP: Inferring regulatory element landscapes and transcription factor networks from cancer methylomes. Genome Biol 2015, 16(1):105.
13. Silva TC, Coetzee SG, Gull N, Yao L, Hazelett DJ, Noushmehr H, Lin DC, Berman BP: ELMER v.2: an R/Bioconductor package to reconstruct gene regulatory networks from DNA methylation and transcriptome profiles. Bioinformatics 2019, 35(11):1974-1977.
14. Zhu Y, Chen Z, Zhang K, Wang M, Medovoy D, Whitaker JW, Ding B, Li N, Zheng L, Wang W: Constructing 3D interaction maps from 1D epigenomes. Nat Commun 2016, 7:10812.
15. Chen Y, Wang Y, Xuan Z, Chen M, Zhang MQ: De novo deciphering three-dimensional chromatin interaction and topological domains by wavelet transformation of epigenetic profiles. Nucleic Acids Res 2016, 44(11):e106.
16. Liu D, Davila-Velderrain J, Zhang Z, Kellis M: Integrative construction of regulatory region networks in 127 human reference epigenomes by matrix factorization. Nucleic Acids Res 2019, 47(14):7235-7246.
17. Cao Q, Anyansi C, Hu X, Xu L, Xiong L, Tang W, Mok MTS, Cheng C, Fan X, Gerstein M et al: Reconstruction of enhancer-target networks in 935 samples of human primary cells, tissues and cell lines. Nat Genet 2017, 49(10):1428-1436.
18. Hait TA, Amar D, Shamir R, Elkon R: FOCS: a novel method for analyzing enhancer and gene activity patterns infers an extensive enhancer-promoter map. Genome Biol 2018, 19(1):56.
19. He B, Chen C, Teng L, Tan K: Global view of enhancer–promoter interactome in human cells. Proceedings of the National Academy of Sciences 2014, 111(21):E2191-E2199.
20. Zhao C, Li X, Hu H: PETModule: a motif module based approach for enhancer target gene prediction. Sci Rep 2016, 6:30043.
21. Roy S, Siahpirani AF, Chasman D, Knaack S, Ay F, Stewart R, Wilson M, Sridharan R: A predictive modeling approach for cell line-specific long-range regulatory interactions. Nucleic Acids Research 2015, 43(18):8694-8712.
22. Whalen S, Truty RM, Pollard KS: Enhancer-promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat Genet 2016, 48(5):488-496.
23. Gao T, Qian J: EAGLE: An algorithm that utilizes a small number of genomic features to predict tissue/cell type-specific enhancer-gene interactions. PLoS Comput Biol 2019, 15(10):e1007436.
24. Talukder A, Saadat S, Li X, Hu H: EPIP: a novel approach for condition-specific enhancer–promoter interaction prediction. Bioinformatics 2019, 35(20):3877-3883.
25. Dzida T, Iqbal M, Charapitsa I, Reid G, Stunnenberg H, Matarese F, Grote K, Honkela A, Rattray M: Predicting stimulation-dependent enhancer-promoter interactions from ChIP-Seq time course data. Peerj 2017, 5.
26. Hafez D, Karabacak A, Krueger S, Hwang YC, Wang LS, Zinzen RP, Ohler U: McEnhancer: predicting gene expression via semi-supervised assignment of enhancers to target genes. Genome Biol 2017, 18(1):199.
27. Zhang Y, Wong CH, Birnbaum RY, Li G, Favaro R, Ngan CY, Lim J, Tai E, Poh HM, Wong E et al: Chromatin connectivity maps reveal dynamic promoter-enhancer long-range associations. Nature 2013, 504(7479):306-310.
28. Li W, Wong WH, Jiang R: DeepTACT: predicting 3D chromatin contacts via bootstrapping deep learning. Nucleic Acids Research 2019, 47(10).
29. Zhang H, Li F, Jia Y, Xu B, Zhang Y, Li X, Zhang Z: Characteristic arrangement of nucleosomes is predictive of chromatin interactions at kilobase resolution. Nucleic Acids Res 2017, 45(22):12739-12751.
30. Al Bkhetan Z, Plewczynski D: Three-dimensional Epigenome Statistical Model: Genome-wide Chromatin Looping Prediction. Sci Rep 2018, 8(1):5217.
31. Al Bkhetan Z, Plewczynski D: Multi-levels 3D Chromatin Interactions Prediction Using Epigenomic Profiles. In: 2017; Cham. Springer International Publishing: 19-28.
32. Belokopytova PS, Nuriddinov MA, Mozheiko EA, Fishman D, Fishman V: Quantitative prediction of enhancer-promoter interactions. Genome Res 2020, 30(1):72-84.
33. Yang Y, Zhang R, Singh S, Ma J: Exploiting sequence-based features for predicting enhancer-promoter interactions. Bioinformatics 2017, 33(14):i252-i260.
34. Singh S, Yang Y, Póczos B, Ma J: Predicting enhancer-promoter interaction from genomic sequence with deep neural networks. Quantitative Biology 2019, 7(2):122-137.
35. Mao W, Kostka D, Chikina M: Modeling Enhancer-Promoter Interactions with Attention-Based Neural Networks. bioRxiv 2017:219667.
36. Zeng W, Wu M, Jiang R: Prediction of enhancer-promoter interactions via natural language processing. BMC Genomics 2018, 19(Suppl 2):84.
37. Gan M, Li W, Jiang R: EnContact: predicting enhancer-enhancer contacts using sequence-based deep learning model. PeerJ 2019, 7:e7657.
38. Zhang R, Wang Y, Yang Y, Zhang Y, Ma J: Predicting CTCF-mediated chromatin loops using CTCF-MP. Bioinformatics 2018, 34(13):i133-i141.
39. Kai Y, Andricovich J, Zeng Z, Zhu J, Tzatsos A, Peng W: Predicting CTCF-mediated chromatin interactions by integrating genomic and epigenomic features. Nat Commun 2018, 9(1):4221.
40. Trieu T, Martinez-Fundichely A, Khurana E: DeepMILO: a deep learning approach to predict the impact of non-coding sequence variants on 3D chromatin structure. Genome Biol 2020, 21(1):79.
41. Hong S, Kim D: Computational characterization of chromatin domain boundary-associated genomic elements. Nucleic Acids Res 2017, 45(18):10403-10414.
42. Gan W, Luo J, Li YZ, Guo JL, Zhu M, Li ML: A computational method to predict topologically associating domain boundaries combining histone Marks and sequence information. BMC Genomics 2019, 20(Suppl 13):980.
43. Sefer E, Kingsford C: Semi-nonparametric modeling of topological domain formation from epigenetic data. Algorithms Mol Biol 2019, 14:4.
44. Huang J, Marco E, Pinello L, Yuan GC: Predicting chromatin organization using histone marks. Genome Biol 2015, 16:162.
45. Schreiber J, Libbrecht M, Bilmes J, Noble WS: Nucleotide sequence and DNaseI sensitivity are predictive of 3D chromatin architecture. bioRxiv 2018:103614.
46. Farre P, Heurteau A, Cuvier O, Emberly E: Dense neural networks for predicting chromatin conformation. BMC Bioinformatics 2018, 19(1):372.
47. Fudenberg G, Kelley DR, Pollard KS: Predicting 3D genome folding from DNA sequence with Akita. Nature Methods 2020, 17(11):1111-1117.
48. Schwessinger R, Gosden M, Downes D, Brown RC, Oudelaar AM, Telenius J, Teh YW, Lunter G, Hughes JR: DeepC: predicting 3D genome folding using megabase-scale transfer learning. Nature Methods 2020, 17(11):1118-1124.
49. Di Pierro M, Cheng RR, Lieberman Aiden E, Wolynes PG, Onuchic JN: De novo prediction of human chromosome structures: Epigenetic marking patterns encode genome architecture. Proc Natl Acad Sci U S A 2017, 114(46):12126-12131.
50. Buckle A, Brackley CA, Boyle S, Marenduzzo D, Gilbert N: Polymer Simulations of Heteromorphic Chromatin Predict the 3D Folding of Complex Genomic Loci. Mol Cell 2018, 72(4):786-797 e711.
51. Qi Y, Zhang B: Predicting three-dimensional genome organization with chromatin states. PLoS Comput Biol 2019, 15(6):e1007024.
52. Al Bkhetan Z, Kadlof M, Kraft A, Plewczynski D: Machine learning polymer models of three-dimensional chromatin organization in human lymphoblastoid cells. Methods 2019, 166:83-90.
53. Moore JE, Pratt HE, Purcaro MJ, Weng Z: A curated benchmark of enhancer-gene interactions for evaluating enhancer-target gene prediction methods. Genome Biol 2020, 21(1):17.
54. Cao F, Fullwood MJ: Inflated performance measures in enhancer-promoter interaction-prediction methods. Nat Genet 2019, 51(8):1196-1198.
55. Cao C, Liu F, Tan H, Song D, Shu W, Li W, Zhou Y, Bo X, Xie Z: Deep Learning and Its Applications in Biomedicine. Genomics Proteomics Bioinformatics 2018, 16(1):17-32.
56. Smemo S, Campos LC, Moskowitz IP, Krieger JE, Pereira AC, Nobrega MA: Regulatory variation in a TBX5 enhancer leads to isolated congenital heart disease. Hum Mol Genet 2012, 21(14):3255-3263.
57. Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T et al: An atlas of active enhancers across human cell types and tissues. Nature 2014, 507(7493):455-461.
58. Bianco S, Lupianez DG, Chiariello AM, Annunziatella C, Kraft K, Schopflin R, Wittler L, Andrey G, Vingron M, Pombo A et al: Polymer physics predicts the effects of structural variants on chromatin architecture. Nat Genet 2018, 50(5):662-667.
59. Moquin SA, Thomas S, Whalen S, Warburton A, Fernandez SG, McBride AA, Pollard KS, Miranda JL: The Epstein-Barr Virus Episome Maneuvers between Nuclear Chromatin Compartments during Reactivation. J Virol 2018, 92(3).
60. Sin-Chan P, Mumal I, Suwal T, Ho B, Fan X, Singh I, Du Y, Lu M, Patel N, Torchia J et al: A C19MC-LIN28A-MYCN Oncogenic Circuit Driven by Hijacked Super-enhancers Is a Distinct Therapeutic Vulnerability in ETMRs: A Lethal Brain Tumor. Cancer Cell 2019, 36(1):51-67.e57.
61. Gu X, Wang L, Boldrup L, Coates PJ, Fahraeus R, Sgaramella N, Wilms T, Nylander K: AP001056.1, A Prognosis-Related Enhancer RNA in Squamous Cell Carcinoma of the Head and Neck. Cancers (Basel) 2019, 11(3).
62. Dong SS, Yao S, Chen YX, Guo Y, Zhang YJ, Niu HM, Hao RH, Shen H, Tian Q, Deng HW et al: Detecting epistasis within chromatin regulatory circuitry reveals CAND2 as a novel susceptibility gene for obesity. Int J Obes (Lond) 2019, 43(3):450-456.
63. Yizhar-Barnea O, Valensisi C, Jayavelu ND, Kishore K, Andrus C, Koffler-Brill T, Ushakov K, Perl K, Noy Y, Bhonker Y et al: DNA methylation dynamics during embryonic development and postnatal maturation of the mouse auditory sensory epithelium. Sci Rep 2018, 8(1):17348.
64. Molineros JE, Singh B, Terao C, Okada Y, Kaplan J, McDaniel B, Akizuki S, Sun C, Webb CF, Looger LL et al: Mechanistic Characterization of RASGRP1 Variants Identifies an hnRNP-K-Regulated Transcriptional Enhancer Contributing to SLE Susceptibility. Front Immunol 2019, 10:1066.
65. Olafsdottir TA, Theodors F, Bjarnadottir K, Bjornsdottir US, Agustsdottir AB, Stefansson OA, Ivarsdottir EV, Sigurdsson JK, Benonisdottir S, Eyjolfsson GI et al: Eighty-eight variants highlight the role of T cell regulation and airway remodeling in asthma pathogenesis. Nat Commun 2020, 11(1):393.
66. Styrkarsdottir U, Stefansson OA, Gunnarsdottir K, Thorleifsson G, Lund SH, Stefansdottir L, Juliusson K, Agustsdottir AB, Zink F, Halldorsson GH et al: GWAS of bone size yields twelve loci that also affect height, BMD, osteoarthritis or fractures. Nat Commun 2019, 10(1):2054.
67. Cheng B, Du Y, Wen Y, Zhao Y, He A, Ding M, Fan Q, Li P, Liu L, Liang X et al: Integrative analysis of genome-wide association study and chromosomal enhancer maps identified brain region related pathways associated with ADHD. Compr Psychiatry 2019, 88:65-69.
68. Gudmundsson J, Sigurdsson JK, Stefansdottir L, Agnarsson BA, Isaksson HJ, Stefansson OA, Gudjonsson SA, Gudbjartsson DF, Masson G, Frigge ML et al: Genome-wide associations for benign prostatic hyperplasia reveal a genetic correlation with serum levels of PSA. Nat Commun 2018, 9(1):4568.
69. Wu C, Pan W: Integration of Enhancer-Promoter Interactions with GWAS Summary Results Identifies Novel Schizophrenia-Associated Genes and Pathways. Genetics 2018, 209(3):699-709.
70. Liu Y, Chang JC, Hon CC, Fukui N, Tanaka N, Zhang Z, Lee MTM, Minoda A: Chromatin accessibility landscape of articular knee cartilage reveals aberrant enhancer regulation in osteoarthritis. Sci Rep 2018, 8(1):15499.
71. Lecellier CH, Wasserman WW, Mathelier A: Human Enhancers Harboring Specific Sequence Composition, Activity, and Genome Organization Are Linked to the Immune Response. Genetics 2018, 209(4):1055-1071.
72. Jiang Y, Qian F, Bai X, Liu Y, Wang Q, Ai B, Han X, Shi S, Zhang J, Li X et al: SEdb: a comprehensive human super-enhancer database. Nucleic Acids Res 2019, 47(D1):D235-d243.
73. Li X, Shi L, Wang Y, Zhong J, Zhao X, Teng H, Shi X, Yang H, Ruan S, Li M et al: OncoBase: a platform for decoding regulatory somatic mutations in human cancers. Nucleic Acids Res 2019, 47(D1):D1044-d1055.
74. Hong H, Jiang S, Li H, Du G, Sun Y, Tao H, Quan C, Zhao C, Li R, Li W et al: DeepHiC: A generative adversarial network for enhancing Hi-C data resolution. PLoS Comput Biol 2020, 16(2):e1007287.
75. Cameron CJ, Dostie J, Blanchette M: HIFI: estimating DNA-DNA interaction frequency from Hi-C data at restriction-fragment resolution. Genome Biol 2020, 21(1):11.
76. Carron L, Morlot JB, Matthys V, Lesne A, Mozziconacci J: Boost-HiC: computational enhancement of long-range contacts in chromosomal contact maps. Bioinformatics 2019, 35(16):2724-2729.
77. Li A, Yin X, Xu B, Wang D, Han J, Wei Y, Deng Y, Xiong Y, Zhang Z: Decoding topologically associating domains with ultra-low resolution Hi-C data by graph structural entropy. Nat Commun 2018, 9(1):3265.
78. Liu Q, Lv H, Jiang R: hicGAN infers super resolution Hi-C data with generative adversarial networks. Bioinformatics 2019, 35(14):i99-i107.
79. Liu T, Wang Z: HiCNN: a very deep convolutional neural network to better enhance the resolution of Hi-C data. Bioinformatics 2019, 35(21):4222-4228.
80. Zhang Y, An L, Xu J, Zhang B, Zheng WJ, Hu M, Tang J, Yue F: Enhancing Hi-C data resolution with deep convolutional neural network HiCPlus. Nat Commun 2018, 9(1):750.
81. Hsieh TS, Fudenberg G, Goloborodko A, Rando OJ: Micro-C XL: assaying chromosome conformation from the nucleosome to the entire genome. Nat Methods 2016, 13(12):1009-1011.
82. Abbas A, He X, Niu J, Zhou B, Zhu G, Ma T, Song J, Gao J, Zhang MQ, Zeng J: Integrating Hi-C and FISH data for modeling of the 3D organization of chromosomes. Nat Commun 2019, 10(1):2049.
83. Kloetgen A, Thandapani P, Ntziachristos P, Ghebrechristos Y, Nomikou S, Lazaris C, Chen X, Hu H, Bakogianni S, Wang J et al: Three-dimensional chromatin landscapes in T cell acute lymphoblastic leukemia. Nat Genet 2020, 52(4):388-400.
