


实验笔记丨CRISPR-dCas9实验步骤详解

CRISPR/Cas9 基因编辑技术的基本原理是,Cas9 蛋白与人工设计的 sgRNA 结合成为 sgRNA-Cas9 蛋白复合体并在其引导之下结合到特定的核苷酸序列切割目标 DNA分子造成双链断裂,细胞内非同源末端连接的方式(NHEJ)造成断裂位点随机插入、删除等突变。CRISPR/Cas9 系统通过这种方式引入特定位点的突变。
实验笔记丨CRISPR-dCas9实验步骤详解
CRISPR/Cas9 基因编辑技术的基本原理是,Cas9 蛋白与人工设计的 sgRNA 结合成为 sgRNA-Cas9 蛋白复合体并在其引导之下结合到特定的核苷酸序列切割目标 DNA分子造成双链断裂,细胞内非同源末端连接的方式(NHEJ)造成断裂位点随机插入、删除等突变。CRISPR/Cas9 系统通过这种方式引入特定位点的突变。
dCas9 是通过抑制Cas9 核酸酶的 RuvC 和 HNH 两个结构域的活性来得到的,在此之后得到的 dCas9 系统只有结合基因组序列的能力却没有在基因组序列上切割的能力。CRISPR /dCas9 系统分别 融 合 一 个 激 活 元 件 或 一 个 抑 制 元 件 时 会 产 生 基 因 激 活 或 者 抑 制 的 功 能 。随着dCas9 发现和应用的成熟,CRISPR-dCas9 系统应运而生,逐渐成为科学家分子生物学用于激活和抑制基因表达研究的新工具。
sgRNA 的设计
sgRNA,单链引导 RNA,作为 CRISPR/Cas9 系统中 cas 蛋白的引导序列,可参照 sgRNA 工具网站 ( http:// crispr.mit.edu ) 设计多条 sgRNA 以降低脱靶率。
sgRNA 质粒的构建方法
1) 用 BbsI 在 37℃条件下酶切载体质粒 pAC1371。
2) 琼脂糖凝胶电泳,切胶用 DNA 片段回收试剂盒回收目的载体片段
3) 引物退火
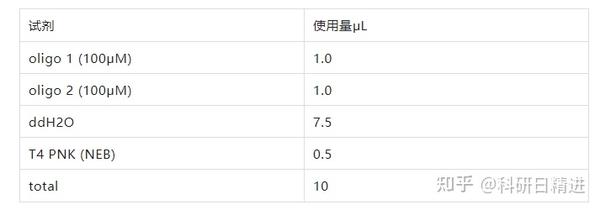
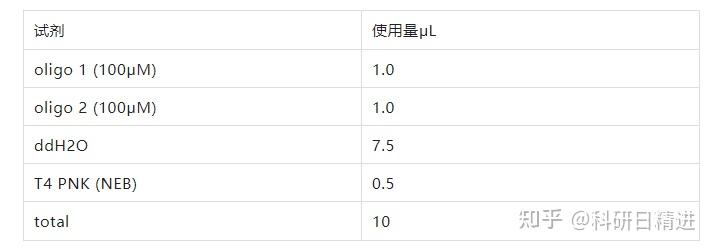
使用以下参数在 PCR 环仪中进行退火:
37℃ 30 分钟,95℃ 5 分钟,然后以每分钟降低 5℃的速度将温度降到 25℃
4) 连接酶连接。
5) 质粒转化(Transformation)
A) 取一支储存在-80℃的 DH5a 感受态细胞放在冰盒里解冻(切不可用手直接握住感受台细胞底部),解冻后将 5-10µL 连接产物加入到感受态细胞中,用枪头轻柔吹混匀(切不可剧烈震荡吹打),在冰盒里静置孵育 25-30 分钟。
B) 冰上孵育后将感受态细胞放在提前调节温度至 42℃水浴锅中热激 90 秒。重新转移冰盒里静置孵育 5-10 分钟。
C) 加入 500μL 提前预热至室温的液体培养基,置于 37℃摇床中复苏摇菌 1 小时。
D) 3000 转离心 3 分钟,弃掉部分上清液,用灭菌冷却至室温的玻璃棒涂板,置于 37℃培养箱中过夜生长。
6) 随机挑取独立生长的菌斑于液体培养基中培养 6-8 小时,进行菌落 PCR。
7) 将 PCR 产物进行琼脂糖凝胶电泳。凝胶成像仪成像,选取阳性菌落送去测序公司进行测序鉴定。
8) 根据测序结果选取目的菌落扩大培养,提取质粒,备后续实验室用。
脂质体转染细胞
1) 准备:细胞准备,将长势良好 HEK293T 细胞按一定数量传代,如:六孔板 1.5*10⁶/孔,传代时培养基需预热。
2) 将长势良好,密度适当的 HEK293T 细胞更换一半新鲜培养基(培养基需要预热,更换培养基时,轻柔操作,不要将贴壁生长的 HEK293T 细胞吹起来)。
3) pMD2.G: 1µg+paspax2: 1µg +transgene: 2µg+optimem: 0.125mL 混匀,室温静置5 分钟, lipofectamine 2000:8µL+optimem: 0.125mL 混匀,室温静置 5 分钟。
4) 5 分钟后将步骤 2 中的两种混合物混合在一起,轻柔混匀,室温静置 15 分钟。
5) 将 3)中混合物加入 6 孔板中,每孔加 250µL(加样时使混合物均匀分布在孔中)。
6) 放入培养箱继续培养。6 小时后,去除并更换 HEK293T 细胞包装培养基 2mL 。
7) 按时用显微镜观察细胞生长状况或者荧光表达情况,并拍照取材。
8) RNA 提取,反转录成 c DNA。Real-time PCR 鉴定基因表达水平。
The surveyor assay of insertion and deletion分析 CRISPR/Cas9 系统在特定位点的作用效率
CRISPR/Cas9 系统在真核细胞中切割会造成 DNA 双链的断裂,我们采用 Dmitry Y.Guschin 等人用来分析 zinc finger nucleases (ZFN)造成的基因突变得方法来分析CRISPR/Cas9 系统作用效率。这种方法依赖于 Surveyor nuclease 的功能。将发生突变位点通过 PCR 将产物量放大,重新形成单链后退火使正常的序列与发生突变的序列形成错配,这种错配可以被 Surveyor nuclease 所识别并切开。通过这种方式可以很清楚的分析 CRISPR/Cas9 系统在特定位点的作用效率。
1) 收集 HEK293T 细胞,1000 转离心 3 分钟,弃尽上清液,加入 PBS 将细胞重悬收集在 1.5mLEP 管内,用 PBS 洗一次。
2) 取适量细胞 1000 转,3 分钟离心,弃尽上清液。
3) 加入 PCR 裂解液(1000 个细胞/1μL PCR Lysis buffer)。
4) 50℃,1 小时;95℃,15 分钟;4℃, 10 分钟。
5) PCR 1.5%琼脂糖凝胶电泳检测 PCR 是否有目的条带。
6) 退火:
3μL PCR 产物
6μL 1xBufferⅡ
95℃,5 分钟,95–85℃at –2◦C/s, 85–25ºCat –0.1℃/s; 4℃, 10 分钟。
7) 将产物放在冰上,加入 0.5μL Surveyor nucLease S 。
PCR 42℃,20 分钟;4℃, 10 分钟。
8) 加入 2μL 10×orange Loading buffer
9) PAGE 胶的配置
将洗干净并烘干的玻璃板用夹子夹住,固定在带有胶垫的架子上。上层胶和下层胶的组分如下:
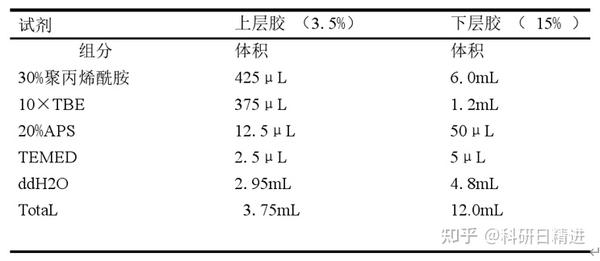
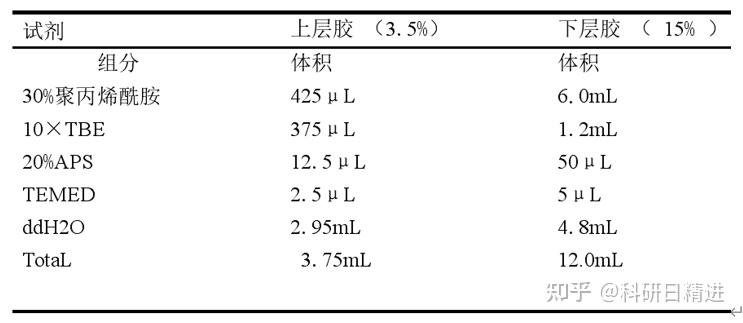
10) 待胶凝固后,放在电泳槽中,加入 1×TBE,80V 电压,预跑 30 分钟。
11) 将 5μL 酶切好的样品点到点样孔中,使用 100bp DNA maker。
12) 80V 电压, 30 分钟; 120V 电压,90 分钟。
13) 把跑好的 PAGE 胶在玻璃板中取下,放在带有 EB 的 1×TBE 中,染色 10 分钟左右。
14) 凝胶成像,利用 ImageJ 软件进行灰度值分析。
PCR 产物送公司测序,进行 TIDE 分析( http:// tide-calculator.nki.nl/ )
其他实验技能分享:
实验笔记丨Far western blotting实验步骤和重点考虑因素
科研日精进:实验笔记丨分子间相互作用分析——微量热泳动(MST)技术介绍
科研日精进:Western Blotting实验中常见问题及解决方案
更多科研实验干货,请关注“科研日精进”VX公众号
