

头图来自2014.11.30的必应壁纸
摸鱼大四人的自白:
毕业设计准备搞这方面的程序优化,完全零基础入门,加之水平一般,一个多月仍然难以成体系,所以整理一下最近学习的知识( 给组会凑点内容 )。

0. 机器学习基础
首先,我们假设同学们已经有一定机器学习基础,熟悉DNN网络的数学原理、搭建、训练和推理。然后,是犯下然后之罪的然后。
1. 分子动力学
分子动力学(Molecular dynamics)是一种计算机辅助模拟工具,用于描述物质或分子中的原子级运动过程[1]。根据研究对象组成原子不同时刻的位置和动量,基于统计力学知识获得想要的物理量,解释对象的性质和行为。其基本过程为:1) 设置研究对象组成原子的初始位置和速度;2) 计算每个原子受到的合力,并基于牛顿第二定律计算原子的加速度;3) 计算原子下一时刻的速度;4) 计算原子下一时刻的位置;5) 循环2) - 4)的过程,得到一系列时刻原子的位置和速度;6) 基于位置和速度信息得到描述对象性质和行为的物理量。
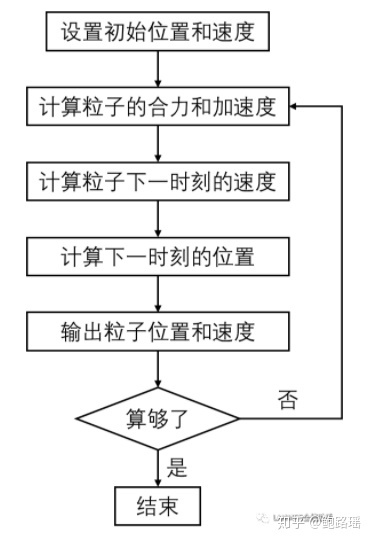
在整个计算过程中,最重要的是如何计算原子间相互作用力,其中包括化学键作用和非键作用,例如静电作用力和范德华力。由于能量关于位移的导函数是力,只要得到原子的势函数,就可以计算出原子此时受到的合外力,同时也可以得到系统总内能。
分子动力学模拟的精度依赖于原子间相互作用的描述方法。其中,基于第一性原理的分子动力学方法(ab initio molecular dynamics),例如density functional theory (DFT),具有突出的精度表现。但其计算开销与电子自由度的3次方成正比,难以应用于原子数量级为10^3-10^8、时间尺度为微秒级的实际应用中。而基于empirical force fields (EFF)的分子动力学方法,虽然能够处理如此大规模的模拟,但其精度较低[1]。
普林斯顿的鄂维南团队在2018年提出一种基于机器学习的分子动力学方法DeePMD[2]。使用由AIMD方法得到的数据训练DNN网络,将训练好的DNN网络作为原子的势函数,提供给LAMMPS等分子动力学模拟软件进行计算。在加快了模拟速度的同时,保证了模拟结果可以达到AIMD方法的精度。结合机器学习的分子动力学是目前很有前景的方向之一,也是我未来一段时间研究的对象,下面将介绍其具体实现的开源软件包DeePMD-kit。
2. DeePMD-kit[3]
2.1 框架
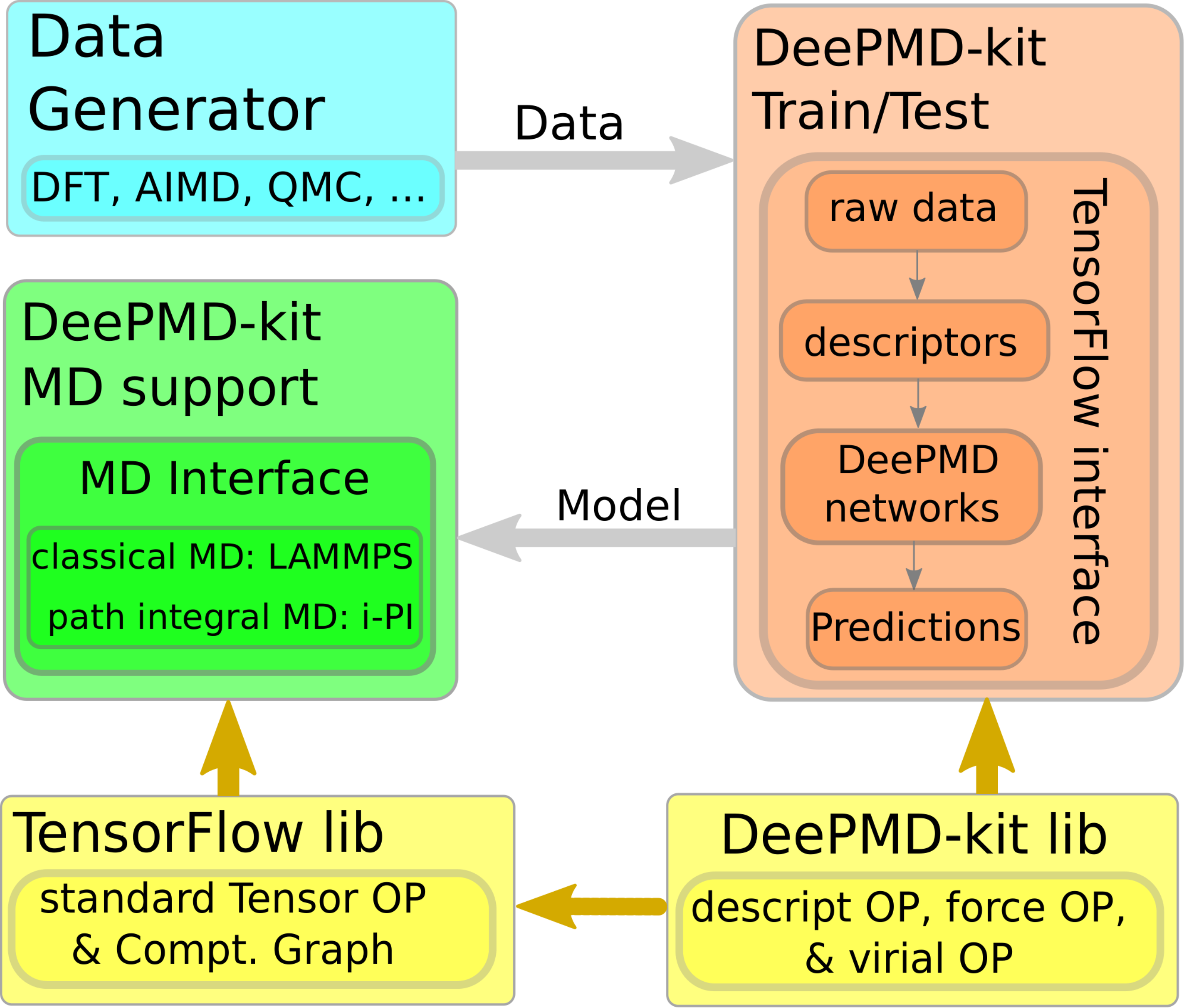
DeePMD-kit包括三部分:1) 用于计算作用力、描述符和作用力的C++库,包括与TensorFlow的接口和第三方MD包;2) 使用TensorFlow的训练和测试程序;3) 对于LAMMPS和i-PI的支持。
一个完整的模拟过程包括以下步骤:1) 对于给定的系统,DeePMD-kit先将AIMD计算得到的数据转化为一种自定义的文件格式,其中包括原子的坐标以及原子的能量、力和virial。2) 随后输入到由TensorFLow框架搭建的势函数网络进行训练,通过原子坐标预测此时的能量。3) 训练好的模型将被冻结保存,提供给融合了DeePMD-kit的传统分子动力学模拟软件,例如LAMMPS。4) 使用LAMMPS对给定的数据进行分子动力学模拟。需要指出的是,对于同一种系统,不需要重新训练势函数模型,可以复用模型,这正是结合机器学习的优势所在。
2.2 原理
对于一个包含N个原子的系统,系统总势能为每个原子的势能之和。
为了简化计算,可以近似认为每个原子的势能由该原子及其截止范围内的邻居原子的坐标决定。
因此考虑使用DNN网络训练一个从原子坐标到其原子势能的函数模型。但这种方法不能体现potential energy surface(PES)中的平移、旋转和置换对称性,因此将原子i的坐标先通过一个DNN网络映射到一个描述符
,再由
通过一个DNN网络映射到能量
[2]。
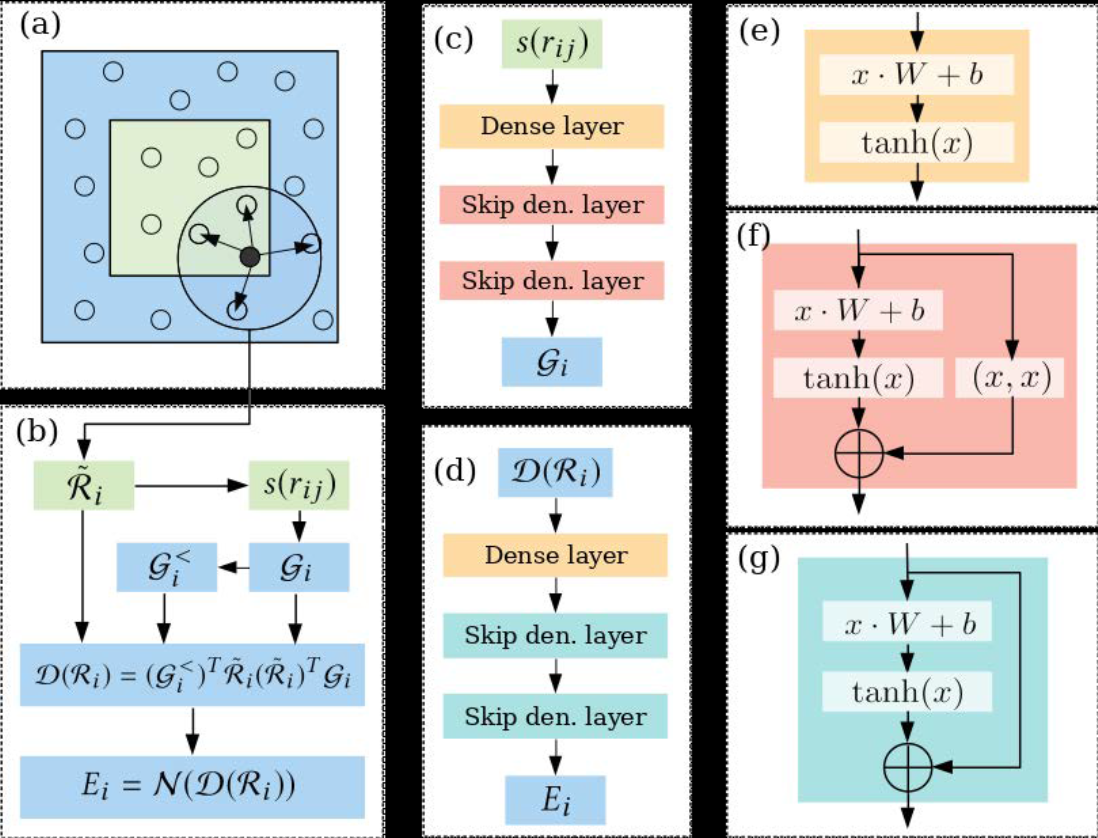
映射中使用的神经网络均为简单的3层DNN网络,并加入了残差模块,如图所示。DeePMD的描述符有多种构造方式,以下介绍最基础的se_e2_a版本描述符
的详细构造过程。
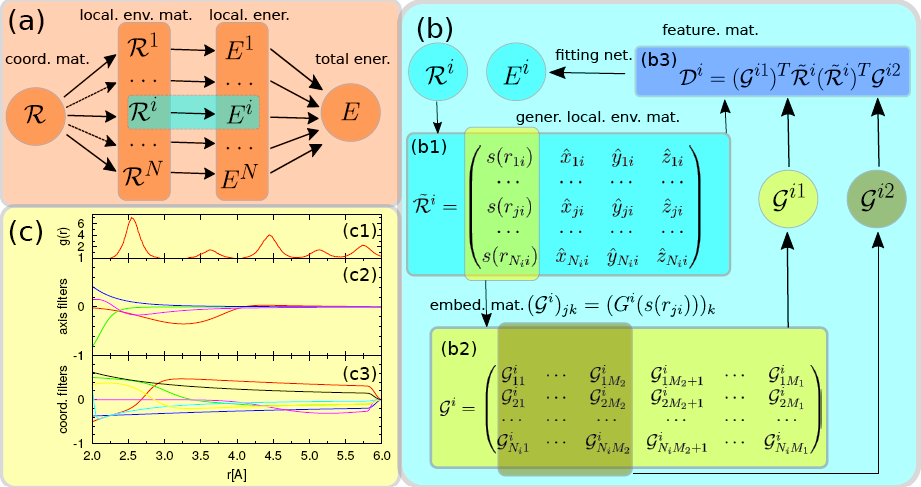
构建描述符的过程如上图(b)所示。对于一个中心原子i,根据其截断半径内的邻居原子,构建矩阵
,行向量为某个邻居原子j与中心原子i的相对坐标。
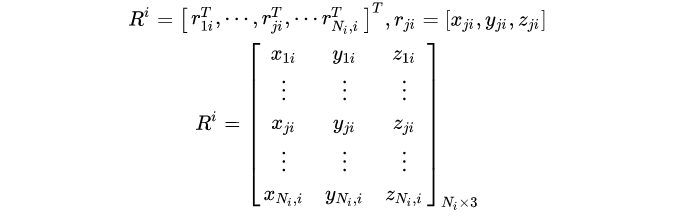
根据自定义的平滑函数s(rji)将其转换为环境矩阵
。
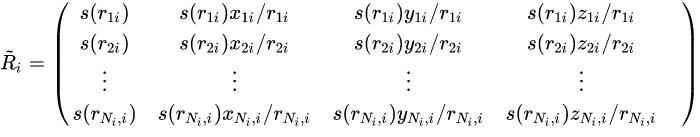
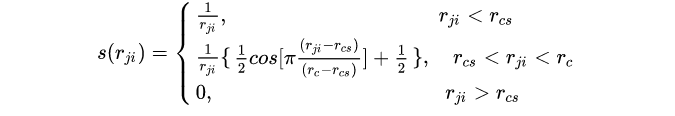
通过DNN网络G得到
行
列的嵌入矩阵
。对于每一个邻居原子的s(rji),网络G将其映射到
宽的行向量。
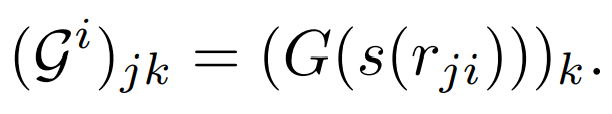
取嵌入矩阵
的前
列,得到矩阵
,
应远小于
。至此,由以下公式可以计算得到描述符
,计算过程中除原子坐标和邻居原子数外均为模型的超参数。该模型的计算复杂度与
成线性关系。
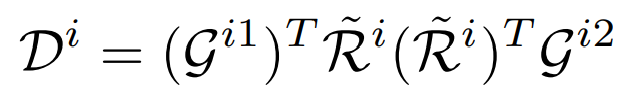
经过一些“简单”的推导,显然可以证明,通过上式构造的描述符满足平移、旋转、置换对称性,满足要求[4]。
2.3 软件[2][3]
2.3.1 代码结构
看不懂代码没关系,我也没看懂。
-
data/raw: tools manipulating the raw data files. -
examples: examples. -
deepmd: DeePMD-kit python modules. -
source/api_cc: source code of DeePMD-kit C++ API. -
source/ipi: source code of i-PI client. -
source/lib: source code of DeePMD-kit library. -
source/lmp: source code of Lammps module. -
source/gmx: source code of Gromacs plugin. -
source/op: tensorflow op implementation. working with library.
2.3.2 数据准备
DeePMD定义了RAW数据协议。数据包括一组systems,每个system(RAW)文件包括数个frames(一行),被划分为训练集和测试集。每个frame记录模拟区域的shape(三维张量)以及system中所有原子的坐标,标注能量、力和virial,任意1项或2项可能缺失,这些标签可以由其他程序计算得出。
2.3.3 模型训练
计算原子能量分为两步:1) 根据相对位置构造出描述符; 2) 将描述符输入DNN。后者使用标准TensorFlow算子,而前者使用C++实现的自定义算子。
2.3.4 模型冻结和测试
2.3.5 分子动力学模拟
使用定制的LAMMPS 和 i-PI进行MD仿真。
3. LAMMPS[5][6]
如果你也想像我一样,(只会)熟练地使用命令行对给定参数和数据进行分子动力学模拟,请参照以下教程食用。
6. Run MD with LAMMPS — DeePMD-kit documentation (https://docs.deepmodeling.org/projects/deepmd/en/master/getting-started/lammps.html)
Lennard jones mixture (https://lammpstutorials.github.io/tutorial01.html)
LAMMPS入门教程(2)——从in文件建立讲分子动力学模拟流程 - 知乎 (https://zhuanlan.zhihu.com/p/341613975)
4. 展望
Pushing the limit of molecular dynamics with ab initio accuracy to 100 million atoms with machine learning.
UCB的贾伟乐博士(现为中科院计算所副研究员)在2020年发表了一篇取得ACM Gordon Bell Prize的论文,标题如上。通过大规模并行的方式,将基于机器学习的分子动力学方法应用于Summit超级计算机上,使其能够支持更大空间和时间尺度上的模拟。
其创新点包括3个部分,算法创新、神经网络创新以及减小MPI通信瓶颈。算法创新包括:1) 通过对邻域列表引入了新的数据布局,避免计算嵌入矩阵的分支,从而提高计算粒度;2) 邻域列表的新数据结构中的元素被压缩到64位整型中,可以更有效地使用GPU优化专门TensorFlow算子;3) DP模型的混合精度计算,使得计算密集型任务使用单精度或半精度而不损失准确度。神经网络创新在于使用通用矩阵乘GEMM算子优化标准TensorFlow算子。减小MPI通信瓶颈的过程中利用了一些物理特性以及高性能计算的优化方法[1]。
我认为,科学计算程序通过结合HPC和AI的方法提升性能,扩展应用范围,大概是未来的热点研究方向之一(
不是也没关系,能让我毕业就行
)。
参考资料
[1] Jia, Weile , et al. "Pushing the limit of molecular dynamics with ab initio accuracy to 100 million atoms with machine learning." 2020.
[2] H. Wang, L. Zhang, J. Han, and W. E, “DeePMD-kit: A deep learning package for many-body potential energy representation and molecular dynamics,” Computer Physics Communications, vol. 228, pp. 178–184, 2018.
[3] deepmodeling/deepmd-kit: A deep learning package for many-body potential energy representation and molecular dynamics (https://github.com/deepmodeling/deepmd-kit)
[4] Zhang, Linfeng , et al. "End-to-end Symmetry Preserving Inter-atomic Potential Energy Model for Finite and Extended Systems." (2018).
[5] LAMMPS Molecular Dynamics Simulator (https://www.lammps.org/)
[6] LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales , A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. in 't Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott, S. J. Plimpton, Comp Phys Comm, 271 (2022) 10817.