


Open Babel 分子格式转换工具

Open Babel(以下简称obabel)是化学领域常用的一个文件格式转换工具,支持xyz、SMILES、InChI、mol以及mol2等格式之间的互相转化。
官方的文档宣称可以支持超过110种化学格式。
写这个文章,主要是想总结一些原来在项目过程中使用obabel的一些场景。
(本文章不涉及安装教程,安装过程较简单的方法是使用conda,可以参阅官方文档)
基本转换命令
obabel直接跟着要转换的分子文件名称
-O选项 为输出文件名称。obabel会自动识别文件扩展名。
#origin.xyz 转换为 transfer.mol2
obabel origin.xyz -O transfer.mol2生成3D构像
生成3D构像一般用于smile等只有2D结构的文件,转换为3D带有空间结构的构像。
选项 --gen3d
obabel origin.smi -O transfer.mol2 --gen3D 文件切分
选项 -m
一般用于被转换的文件中有多个分子。转换后文件将这些分子单独保存
obabel origin.smi -O transfer.mol2 -m切分后的分子文件名字以-O参数为基准,
例如上面这个命令,转换后的mol2文件会以transfer 1 .mol2 、transfer 2. mol2、transfer 3. mol2 。。。进行重命名。
去除重复分子
选项 --unique
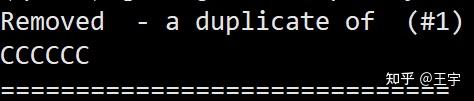
在做虚拟筛选过程中,不同商业库可能包含同一种分子,(同一个分子,不同代理商都在卖), 做虚拟筛选的结果可能有重复的分子存在。所以需要去除重复
当obabel发现重复分子就会删除这个分子。
合并文件
选项 -j / --join
obabel 后跟着要合并的文件,最后加一个-j选项
obabel protein.pdb ligand.mol2 -O combine.pdb -j在原来我写过一篇文章中提到过的 PLIP 分析相互作用:
PLIP 需要的是复合物文件,而大部分分子对接程序得到的结果,都是一个个配体文件,没有与蛋白质成为复合物。
obabel这个小功能则省去了很多麻烦。写一个Python脚本,利用 os.system() 调用obabel就可以循环的生成虚拟筛选得到的蛋白质-配体复合物文件。
但这种合并在实际使用过程中Bug还是偶尔会碰到。
加氢(谨慎使用)
选项 -h 表示加全氢,不考虑质子化状态
选项 -p <pH> 表示加指定pH的氢
慎重使用,此方法加氢可能与专业软件所算的质子化状态不同(例如Schrodinger),
有版权的建议使用其LigPrep模块。
不建议使用obabel转换pdbqt
在使用vina进行虚拟筛选,配体的格式需要转换为pdbqt。
而obabel转换的pdbqt不能用,原子没有带电描述。
建议直接使用prepare_ligand.py脚本循环转换。可以参考使用Gypsum-DL
在转换格式的工具中,obabel本身很强大,但是很多情况下总是不尽人意。
例如小分子拆分,转换之后并不能以独立的分子名称重命名。RDkit获得分子的属性并输出会更好一点。
在输出pdb格式也是有一定的bug,Biopython处理的会更好一点。
在实验和项目过程中,学会多种工具配合使用。
最近接连被裁员处于失业中,身体欠佳。打算回东北老家卖烤冷面去了。
有时间我会把先前做的项目和使用过好用的工具给大家写一写。有什么问题也可以私信,不过不保证能及时回复哈。希望能帮到每个在CADD入坑的小伙伴。

